- *Corresponding Author:
- R. P. R. Monica
Department of Pharmaceutics, AISSMS College of Pharmacy, Pune-411 001, India
E-mail: monicarp_6@hotmail.com
| Date of Submission | 09 August 2016 |
| Date of Revision | 15 December 2016 |
| Date of Acceptance | 09 March 2017 |
| Indian J Pharm Sci 2017;79(2):241-249 |
Abstract
Microporous osmotic tablets were developed using controlled porosity membrane, which delivers drug in a controlled manner for prolonged period of time. The objective of the present investigation was to formulate and evaluate an oral osmotic drug delivery system for the antidepressant drug venlafaxine hydrochloride to achieve zero order release. Venlafaxine hydrochloride was selected as it has a short biological half-life and high aqueous solubility. Drug-excipient compatibility was studied using Fourier transform infrared spectroscopy. Core tablets were prepared by wet granulation method followed by coating. Sodium chloride and potassium chloride (1:1) were used as osmotic agents in the core tablet and polymers Eudragit RLPO and RSPO (1:1) were used for coating along with sorbitol (70% w/w) as pore formers. The tablets showed desired sustained release profile for 24 h. Optimization studies were performed to study the effect of concentration of osmotic agent and pore former on drug release (t50% and t90%). From in vitro release studies, it was evident that drug release was independent of dissolution media, agitation intensity, but highly dependent on the concentration of pore forming agents, osmotic agent and weight gain of the tablet coating. Scanning electron microscopy confirmed the microporous structure of optimized batch. The optimized formulation gave desired once a day release of venlafaxine hydrochloride without using laser drilling technique which would make it more patient compliant and cost effective.
Keywords
Venlafaxine hydrochloride, microporous osmotic tablet, sorbitol, sodium chloride, potassium chloride, osmotic pressure
The purpose of any drug delivery system is to target the required amount of drug to the site of action and to maintain the desired plasma drug concentration [1]. Conventional oral drug delivery systems supply an instantaneous dose of drug, which cannot control the release and effective concentration of drug at the target site [2].
Osmotically controlled oral drug delivery systems utilize osmotic pressure for controlled delivery of active agents [3]. These systems can be utilized for systemic as well as targeted delivery of drugs. The release of drugs from osmotic system is governed by various formulation factors such as the solubility and the osmotic pressure of the core components, nature of the rate controlling membrane [4]. Drugs can be delivered in a controlled pattern over a long period of time by the process of osmosis. Osmotic devices like Rose-Nelson pump, Higuchi-Theeuwes pump and elementary osmotic pump are the most promising systems for controlled drug delivery [5,6].
Osmotic technology has been investigated to obtain controlled release profile for drugs such as theophylline, salbutamol sulphate [7], metoprolol tartrate [8] and diltiazem hydrochloride [9]. Swellable elementary osmotic pump was developed to achieve zero-order release for nifedipine, which is poorly water-soluble [10]. Park et al. [11] developed a novel squeeze-type osmotic tablet for nifedipine composed of one or more polymeric ring type of squeeze-push layer (squeeze-disc) and a centered drug core. An assymetric membrane system was developed for felodipine and nifedipine to improve the osmotic effect and achieve controlled release of the model drugs [12].
Venlafaxine hydrochloride (VH), an antidepressant, is a Biopharmaceutical Classification System (BCS) class 1 drug with high aqueous solubility and bioavailability close to 1. However, it has a relatively short elimination half-life of 5 h, thereby requiring twice a day dosing, which often leads to patient non-compliance [13]. Thus, there is a strong clinical need and market potential for a dosage form that will deliver VH in a controlled manner to enhance the patient compliance. Matrixbased systems have been developed for VH, which showed anomalous non-Fickian release profile [14,15]. Sandhya et al. developed a controlled porosity osmotic pump tablet using swellable polymers to achieve a first order release profile of 8 h duration [16]. The present study was aimed towards the development of an extended release once a day formulation of VH based on osmotic technology. Different formulation variables were studied and optimized to achieve a zero-order release profile.
Materials and Methods
VH was obtained from Avtos Life Sciences, Mumbai. Eudragit was obtained from Evonik Industries, Essen, Germany. Sodium chloride (NaCl), potassium chloride (KCl), mannitol, lactose, sorbitol were obtained from Loba Chemie Ltd, Mumbai and acetone, PEG-400, PVP K30, magnesium stearate, talc were obtained from S. D. Fine-Chem Limited, Mumbai.
Preliminary studies:
VH microporous osmotic tablets were prepared in two steps. Four trial batches of core tablets were prepared using various electrolyte osmotic agents such as NaCl, KCl and non-electrolyte osmotic agents such as mannitol, singly and in combination. These batches were subsequently coated with combination of Eudragit RLPO and Eudragit RSPO [17], in various concentrations up to 3%. Coating solutions with different pore forming agents like sorbitol (70% w/w) and sucrose were prepared. Each of these coating solutions contained polyethylene glycol 400 (PEG 400) and dibutyl phthalate (DBP), singly and in combination as plasticizers. Coating of the core tablet was done so as to achieve 3, 5 and 7% weight gain [18].
Compatibility studies:
Physical mixtures of drug and excipients were prepared to study the compatibility, which was carried out using FTIR-8300, Shimadzu Co., Kyoto, Japan by potassium bromide (KBr) pellet method. Samples were mixed with KBr in 1:100 proportion and spectra was recorded in range of 4000-400 cm-1. These samples were subjected to compatibility studies and stored for 30 d at 40 ± 2○/75 ± 5% relative humidity in glass vials. IR spectra of these stored samples were then obtained after 30 d.
Experimental design:
In the present study 32 full factorial design was employed to study the effect of selected variables and was carried out with Design‐Expert software 9 (Stat‐Ease Inc, Minneapolis, USA). The concentration of NaCl:KCl (1:1) and sorbitol (70% w/w) considered as percent weight of polymer were selected as the independent variables while time for 50% drug release (t50%) and time for 90% drug release (t90%) drug release were selected as the dependent variables. These two timepoints of drug release were calculated by subjecting the in vitro release data to DD Solver software. Each independent variable was investigated at three levels (i.e.-1, 0, +1) as given in Table 1. The observed and predicted responses were critically compared. The optimized formulation was selected on the basis of desirability based acceptance criteria and percentage error. The effect of various formulation variables was studied on the optimized batch [19].
| Batch | Factor A | Factor B |
|---|---|---|
| NaCl:KCl (1:1) | Sorbitol (70% w/w)% w/w of Eudragits | |
| % w/w of tablet | ||
| 1 | 30(-1) | 15(-1) |
| 2 | 35(0) | 15(-1) |
| 3 | 40(1) | 15(-1) |
| 4 | 30(-1) | 20(0) |
| 5 | 35(0) | 20(0) |
| 6 | 40(1) | 20(0) |
| 7 | 30(-1) | 25(1) |
| 8 | 35(0) | 25(1) |
| 9 | 40(1) | 25(1) |
Table 1: Levels of independent variables for factorial design
Preparation of core tablets:
Granules were prepared by wet granulation method using PVP K30 as a binder; the dried granules were passed through sieve no.16# and blended with talc and magnesium stearate. The homogeneous blend was then compressed into tablets (220 mg each) using 8 mm diameter, deep concave punches in a Rimek mini press II compression machine. The compression force was adjusted to give tablets with hardness of approximately 5-6 kg/cm2 on a Monsanto tablet hardness tester [20].
Evaluation of core tablets:
Tablets were selected randomly from each batch for weight variation, hardness, thickness, friability and assay. Hardness of tablet was measured using a Monsanto tablet hardness tester and thickness was measured by using a Vernier caliper. Friability of tablets was determined on a Roche Friabilator, which was operated for 4 min at 25 rpm. Tablets were dedusted and reweighed and percent friability was calculated [21] using Eqn. 1, % friability = (1-w/w0)×100, where, w0 is the initial weight of tablets, w is the weight of tablets after friability testing. Content uniformity of core tablets was determined for each formulation. The core tablets were crushed and powder equivalent to 10 mg was weighed and dissolved in 100 ml methanol in a volumetric flask and sonicated for 30 min. Absorbance was measured on a UV double beam spectrophotometer (UV 3000 LabIndia) at 224 nm against methanol as blank after suitable dilutions [22].
Coating of core tablet:
For coating solution, required amount of Eudragit RLPO and RSPO were dissolved in sufficient amount of isopropyl alcohol:acetone (30:70) at 40° in a thermostat water bath. To this solution, appropriate amount of plasticizers like PEG400 and dibutylpthalate were added. Aqueous pore forming agent (70% w/w sorbitol) was dispersed in methanol and added to solution of Eudragit polymers with sunset yellow as coloring agent. A fine dispersion was formed, which was then shaken manually for 10-15 min to uniformly disperse the pore forming agent and coloring agent. The coating was carried out in the Pharma R and D coater (Ideal Cures Pvt. Ltd) for 25 tablets with pan speed of 25 rpm, pan temperature of 60-65°, atomizing air pressure of 5-6 psi and spray rate of 1 ml/min. The tablets were coated till 5% weight gain was obtained. Coated tablets were further air dried for 24 h.
In vitro drug release:
USP type II (paddle type) dissolution apparatus (TDT-08l, Electrolab, Mumbai, India) were used for dissolution studies in 900 ml medium at 37 ± 0.5° at a rotation speed of 50 rpm [23]. All nine batches of tablets were transferred to the dissolution medium containing 900 ml of 0.1 N HCl for first 2 h followed by phosphate buffer pH 6.8 for remaining 22 h. The dual pH dissolution medium was used to simulate physiological conditions as specified in Indian Pharmacopoeia (IP) 2014 [24] for sustained release preparations. At 1 h interval, aliquots of 1 ml were withdrawn, filtered and absorbance was measured by UV spectrophotometer at 224 nm [25]. The medium was replaced with equal volume of fresh dissolution medium to maintain sink conditions.
Effect of agitation intensity and osmotic pressure of dissolution medium:
The release profile of optimized formulation in 900 ml dissolution media was studied separately. Dissolution media was used as previously described and dissolution apparatus was operated at speeds of 50, 75 and 100 rpm. For studying the effect of osmotic pressure on release profile, dissolution media of different osmotic pressures were prepared using different strengths of urea (0 M, 4 M and 8 M) with stirring speed of 50 rpm [19,23].
Scanning electron microscopy (SEM):
In order to evaluate the structural changes of the semi permeable surface, SEM images of the coated tablets were obtained before and after dissolution test using scanning electron microscope (XL30 ESEM, Philips, Eindhoven, Netherlands). The tablets (coated tablets before dissolution studies) were mounted as such on the specimen stub. On the other hand, small sample of the coating membrane was carefully cut from the exhausted shells (after 24 h of dissolution studies) and dried at 50° for 12 h. The mounted samples were sputter coated for 5 to 10 min with gold using fine coat ion sputter and examined under SEM [26,27].
Pharmacodynamic studies:
Approval to carry out in vivo study was obtained from Institutional Animal Ethics Committee (IAEC) of AISSMS College of Pharmacy and Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA) guidelines were followed for the studies. The optimized tablet was subjected to these studies using Swiss albino mice as the animal model. The animals were divided into two sets of three groups and each group comprised of six mice. In each set one group was control group whereas one group was dosed with marketed sustained release tablet, Ventab, (Intas Pharmaceuticals Ltd.) and third group was dosed with the optimized osmotic tablet. Two pharmacodynamic models were selected for the study.
Despair swim test in mice:
The mice, weighing 30-50 g was used were individually forced to swim inside a vertical Plexiglas cylinder fabricated as for Porsolt forced swim chamber [28]. Containing water maintained at 25°. The mice were placed in the chambers and observed for the vigour in their swimming initially followed by periods of immobility before and after administration of the test and marketed tablets at time points of 4, 8, 12, 16, 20 and 24 h. The animals were transferred to their cages after observation at each time point [29].
Tail suspension test in mice:
The mice, weighing 20-25 g were housed in their cages for at least 10 d prior to testing in a 12 h light cycle with freely available food and water. Groups of 6 animals were treated with the test compounds or the vehicle orally 60 min prior to testing. For the test, mice were suspended above a solid but cushioned surface at 58 cm using medical adhesive tape approximately 1 cm from the tip of the tail. The duration of immobility was recorded for periods of 5 min. Mice were considered immobile when they hung passively and completely motionless for at least 1 min [30].
Stability studies:
The optimized formulation was subjected to accelerated stability studies as per the International Conference of Harmonization (ICH) guidelines [22]. The optimized formulation was sealed in an aluminum foil and stored at 40 ± 2° and 75% relative humidity for 3 mon. Tablets were removed at 30, 60 and 90 d and evaluated for physical characteristics and in vitro drug release [31].
Results and Discussion
Preliminary studies were conducted for selecting the osmogens for core tablet, pore formers and plasticizers. The tablets containing different osmogens were coated with a combination of Eudragit RLPO and RSPO coating polymers in 1:1 ratio with varying amounts of sorbitol (70% w/w) or sucrose as pore formers in strengths of 20 and 30% w/w. Dissolution studies in USP type II dissolution apparatus using 0.1 N HCl for 2 h and phosphate buffer pH 6.8 for 22 h, were carried out on the preliminary batches. Tablets with combination of NaCl and KCl as osmogens in 1:1 ratio had a t50%of 11.6 h whereas tablets with single electrolyte osmogen had a t50% of 7.72 h (NaCl) and 7.99 h (KCl). The t50% for tablets containing mannitol as osmogen was around 9 h. The coating prepared with Eudragit polymers containing sucrose as pore former and PEG400 as plasticizer was found to rupture in 14 h leading to complete release of drug. It has been reported that high levels of certain pore formers may lead to weakened films that could burst prematurely [32].
Various plasticizers that were studied included PEG400 and DBP; alone and in combination. It was found that both the plasticizers when used singly released significantly higher or lower amount of drug whereas their combination gave required drug release for achieving the target of once a day dosing. Tablet coatings with PEG400, which is highly hydrophilic gave faster drug release in a shorter timespan while DBP, which being hydrophobic, gave much lower release in longer timespans. Considering all these factors further experiments were conducted using NaCl and KCl as osmogens (1:1), sorbitol as pore former and DBP as plasticizer with 5% weight gain of tablet.
Compatibility studies for VH were carried out with potential formulation excipients to determine possibility of drug-excipient interaction/incompatibility. FTIR analysis did not show any evidence of adverse interaction between drug and excipients. A minor shifting of peak or decrease in peak intensity could be attributed to dilution effect in the mixture.
A 32 factorial design was used to study the effect of concentration of NaCl:KCl (1:1) and sorbitol (70% w/w) on time for 50% drug release (t50%) and time for 90% drug release (t90% were selected as the dependent variables. The data generated is given in Table 1, which was analyzed using Design-Expert software 9.0 and polynomial equations were obtained for the same. The experimental values obtained were compared with those predicted by the mathematical models. The interactions between the factors were demonstrated using 3D response surface graphs.
The polynomial equations were found to have high statistical significance (P<0.001) as determined by ANOVA. The fitted full equations relating the responses of t50% (Y1) and t90% (Y2) to the transformed factors are expressed in Eqn. 2: Y1= t50% = 12.09–0.80A–0.79B– 0.34AB–0.050A2+0.32B2.
In this case A, B, A2 were found to be significant model terms. Factor A is concentration of NaCl and KCl in 1:1 ratio and factor B concentration of sorbitol (70% w/w). From the Eqn., it is evident that as the concentration of both the factors increased, time for 50% drug release decreased, which was evident by negative sign with both factors. The negative and low coefficient for term AB for combined effect of both factors indicated an insignificant effect on responses. The higher value of coefficients indicated that the effect of concentration of NaCl:KCl (1:1) was greater as compared to concentration of sorbitol.
The Model F-value of 108.93 implied that quadratic model was significant. The "Pred R-Squared" of 0.9498 was in reasonable agreement with the "Adj R-Squared" of 0.9858; i.e. the difference is <0.2. Eqn. 3: Y2 = t90% =21.77–1.44A–1.43B–0.60AB–0.090A2+0.58B2. In this case A, B, A2 were found to be significant model terms. It is evident that as the concentration of both the factors increased the time for 90% drug release decreased, which was evident by negative sign with both factors. The negative and low coefficient for term AB for combined effect of both factors indicates an insignificant effect on responses. The higher value of coefficients indicated that the effect of concentration of osmogens was greater as compared to concentration of sorbitol.
In this case A, B were found to be significant model terms. From the equation, it is clear that as the concentration of both the factor increases the time for 90% drug release decreased, which was evident by negative sign with both factors. The coefficients indicated that the effect of concentration of osmogens was greater as compared to concentration of sorbitol. The Model F value of 112.37 implied that quadratic model was significant. The "Pred R-Squared" of 0.9499 was in reasonable agreement with the "Adj R-Squared" of 0.9858; i.e. the difference is less than 0.2. Since the values of R2 were quite high for both types of responses i.e. 0.9947 for 50% drug release and 0.9947 for 90% drug release, we may conclude that the polynomial equations form excellent fits to the experimental data and displayed high statistical validity.
The response surface plot, relationship between the variables at different levels and responses was further elucidated by constructing 3D response surface plots. Figures 1 and 2 show the 3D response surface plot for percent drug release. It was observed that as the concentration of osmotic agent increased the t50% and t90% was found to decrease, which could be due to the stronger osmotic pressure, which is a critical driving force for drug release.

Figure 1: Response surface for t50%

Figure 2: Response surface for t90%
The osmotic pressures of all formulations were calculated using the Van’t Hoff equation [33]. The volume of fluid present in each tablet was calculated considering the tablets to be cylindrical compacts. The molar concentration of osmogens in each tablet was calculated to compute the osmotic pressure exerted by the osmogens, which is a key driving factor for release of drug. The contribution of drug and other soluble excipients to the osmotic pressure was not taken into account as this quantity was same in all formulations. Thus the tablets of formulations F1, F4 and F7 had osmotic pressure of 66.996 osmoles, formulations F2, F5 and F8 had osmotic pressure of 78.141 osmoles while formulations F3, F6 and F9 had osmotic pressure equivalent to 89.296 osmoles given in Table 2. It is evident from the t50% data that drug release was faster in formulations with higher osmotic pressure. However, the concentration of pore former also influenced the drug release, with higher concentration giving faster release at higher osmotic pressure than that at lower concentration of pore former at higher osmotic pressure.
| Run | Factor A | Factor B | Response Y1 | Response Y2 | Osmotic pressure |
|---|---|---|---|---|---|
| NaCl:KCl(1:1) % w/w of tablet | Sorbitol (70% w/w) %w/w of Eudragits |
t50% (h) |
t90% (h) |
π (osmoles) | |
| F1 | 30 | 15 | 13.538 ± 1.21 | 24.368 ± 1.22 | 66.996 |
| F2 | 35 | 15 | 13.311 ± 0.94 | 23.960 ± 0.89 | 78.141 |
| F3 | 40 | 15 | 12.672 ± 1.38 | 22.810 ± 1.29 | 89.296 |
| F4 | 30 | 20 | 12.954 ± 0.51 | 23.317 ± 0.48 | 66.996 |
| F5 | 35 | 20 | 11.986 ± 1.41 | 21.575 ± 1.12 | 78.141 |
| F6 | 40 | 20 | 11.238 ± 0.98 | 20.229 ± 0.96 | 89.296 |
| F7 | 30 | 25 | 12.672 ± 1.12 | 22.810 ± 1.01 | 66.996 |
| F8 | 35 | 25 | 11.622 ± 1.78 | 20.920 ± 1.56 | 78.141 |
| F9 | 40 | 25 | 10.466 ± 1.68 | 18.839 ± 1.48 | 89.296 |
Table 2: Response variables of factorial design experiments (N=3)
Validation of response surface methodology based on the input limits for achieving desired target of drug release, the Design-Expert software 9.0 generated an optimized batch, which was selected based on its desirability (Table 3) and it was further evaluated to obtain the observed values. The optimized tablets, comprising 35.57% of osmogens and 20% sorbitol as pore former were evaluated for release profile. The t50% drug release was computed to be 12.0021 h and t90% was 21.6670 h.
| Formulation no. | Predicted response | Observed response | %Error | Desirability | |||
|---|---|---|---|---|---|---|---|
| t50% | t90% | t50% | t90% | t50% | t90% | ||
| X | 12.0924 h | 21.7667 h | 12.0021 h | 21.6670 h | 0.746 | 0.458 | 0.98 |
Table 3: Validation of response surface methodology
Hardness of the tablets of each batch is in range of 4.00 to 6.00 kg/cm2, which was within IP limits. Thickness of tablets of each batch was in the range of 2 to 3.5 mm, drug content was found to be in the range of 97-99% and friability in range of 0.40-0.55. The average weight of core tablets was approximately 350 mg whereas the coated tablets had average weights in the range of 360 to 375 mg depending on the weight gain of the tablets.
In vitro dissolution studies were performed in order to understand the mechanism and kinetics of drug release. The results of the in vitro drug release were analyzed for various drug release models. These studies were performed by using DD-Solver software shown in Figure 3. In porous osmotic pump tablets the drug release rate depends on the concentration of the osmotic agent and the pore former. Increase in the concentration of osmogen will lead to an increase in the osmotic pressure in the tablet. This causes ingress of water into the core compartment from the surrounding environment across the membrane and causes dissolution of the drug. This will subsequently lead to faster release of the drug [34]. Increase in concentration of pore former resulted in an obvious increase in number of pores and pore size, causing the drug to diffuse out easily. All formulations showed zero order release with value of release exponent ranging from 0.9762-0.9997 and K value (zero order release constant) ranging from 3.693- 4.777.

Figure 3: Dissolution of microporous osmotic tablet of VH 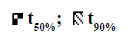
Surface morphology of the coat before and after dissolution studies was studied by SEM at 600X magnification in Figure 4. The micrographs show an intact surface before dissolution and pores of diameters in the range of 10-50 μm in the coating membrane after dissolution studies as shown in Figure 4. These may be forming immediately due to the affinity dissolution of the pore former, which facilitates the inflow of media into the tablet to dissolve the tablet components and generate the osmotic pressure. Eudragit RSPO is insoluble in aqueous media and has low permeability while Eudragit RLPO is insoluble and has high permeability [35]. Both polymers exhibit pH independent swelling. The difference in the permeability’s of the two Eudragits is due to the difference in proportion of quaternary ammonium groups [36]. The Eudragits, being insoluble may be presumed to remain intact (no erosion) during the entire duration of the studies.

Figure 4: SEM Micrographs of the coat, before dissolution and
after dissolution
Scanning electron micrographs of the coat, a) before dissolution
(0 h) and b) after dissolution (24 h)
Effect of various factors on in vitro release from optimized batch was studied, which included agitation intensity and osmotic pressure. Agitation intensities of 50, 75 and 100 rpm was not found to have any influence on percent drug release [19]. Thus the fluid in different parts of the gastrointestinal (GI) tract will hardly affect drug release from the osmotic system. It was independent of the agitational intensity of the release media. The effect of osmotic pressure of surrounding media on drug release was studied in dissolution media containing 4 M urea (osmotic pressure: 101.6 osmoles) and in 8 M urea (osmotic pressure: 203.36 osmoles). The drug release was found to decrease with increase in external osmotic pressure with the optimized formulation displaying a t50% of 15 h in media with lower external osmotic pressure (4 M urea) and of 19 h in media with higher external osmotic pressure (8 M urea). The internal osmotic pressure of optimized batch was found to be 79.422 osmoles. The general mechanism of drug release from osmotic systems involves diffusion of GI fluid across semipermeable membrane at controlled rate causing dissolution of osmogens and other components. As the concentration of dissolved molecules increases the osmotic pressure within the tablet core also increases. The semipermeable membrane is rigid and therefore to reduce osmotic pressure the saturated drug solution is emitted. The rate of entry of biological fluid across the semipermeable membrane into the tablet core will directly affect the rate of drug dissolution and may be defined using the following Eqn. 4 [37]: Dv/dt = A/h Lp (σΔπ-Δρ), where, Dv/dt is the rate of water influx into the tablet core, A is the surface area of the semipermeable membrane, h is the thickness of the semipermeable membrane, Lp is the diameter of the orifice, σ is the reflection coefficient, Δπ is the osmotic pressure difference between the interior of the tablet and the surrounding biological fluids and Δρ is the hydrodynamic pressure difference between interior of the tablet and the surrounding biological fluids.
The drug release rate was found to decrease with increase in osmotic pressure of the media as can be corroborated by above equation, which shows that rate of ingress of water into the tablet is directly proportional to difference in osmotic pressures inside and outside the tablet. However the rate of diffusion of drug to the outside environment is decreased due to the high osmotic pressure of surrounding fluids. In reality the osmotic pressure in the drug delivery system is significantly higher than that of biological fluids and hence this parameter may not be important to understand in vivo performance of the tablets [38].
Pharmacodynamic studies of the tail-suspension test in mice are a behavioural test commonly used for screening antidepressant drugs wherein the escape oriented behaviours are quantified [28]. The forced swim test is a rodent behavioural test wherein the mice are placed in an inescapable transparent tank filled with water and their escape related mobility behaviour is measured [29]. The mice were initially highly active, vigorously swimming in circles, trying to climb the wall or diving to the bottom for 2-3 min interspersed with phases of immobility or floating of increasing length. These periods of immobility were measured. Similar behaviour was quantified in tail suspension test [39]. In all the animals subjected to optimized controlled porosity osmotic tablets, the periods of immobility was found to be lower than in control group and marketed tablet at all-time points under study, which were given in Figures. 5 and 6. The Tukey-Kramer multiple comparisons test [40] was applied to evaluate the statistical significance of the results using Instat software version 3.10. The Q values of the test groups were compared with the q value from the studentized range distribution and were found to be greater than standard value of 3.674 for all time points at P<0.005 with 95% confidence interval.

Figure 5: Forced swim test
║ 4 h mean; ▓ 8 h mean; 12 h mean; 16 h mean; ░ 20 h
mean; 24 h mean

Figure 6: Tail suspension test
║4 h mean; ▓ 8 h mean; ░ 12 h mean; // 16 h mean; ■ 20 h
mean; ═ 24 h mean
The purpose of stability studies are to provide evidence on the quality of a drug substance or drug product, which varies with time under the influence of a variety of environmental factors such as temperature, humidity and light. The stability studies of the optimized osmotic pump tablets revealed no significant changes in the physical parameters and insignificant reduction in the content of the active drug over a period of three months. However, storage temperature not exceeding 45o and moisture proof packaging are essential to ensure stability of these formulations.
Conflict of interests
Authors declare no conflict of interests.
Financial support and sponsorship
Nil.
References
- Qiu Y, Zhang G. Research and development aspects of oral controlled-release dosage forms. Handbook of Pharmaceutical Controlled Release Technology. New York: Marcel Dekker, Inc; 2000. p. 465-503.
- Ratnaparkhi MP, Gupta JP. Sustained release oral drug delivery system: an overview. Int J Pham Res Review 2013;2:11-21.
- Kalakota VR, Kondeti RR. A review on the novel approach to osmotic pump drug delivery system. Int J Innovative Pharm Sci Res 2014;2:1928-42.
- Gupta R, Gupta R, Rathore G. Osmotically controlled drug delivery systems-a review. Int J Pharm Sci 2009;1:269-75.
- Chien YW. Novel drug delivery systems. 2nd ed. New York: Marcel Dekker; 2005. p. 139‐96.
- Makhija SN, Vavia PR. Controlled porosity osmotic pump‐based controlled release systems of pseudoephedrine I, cellulose acetate as a semi permeable membrane. J Control Release 2003;89:5‐18.
- Prabakaran D, Singh P, Kanaujia P, Jaganathan KS, Rawat A, Vyas SP. Modified push-pull osmotic system for simultaneous delivery of theophylline and salbutamol: development and in vitrocharacterization. Int J Pharm 2004;284:95-108.
- Kumaravelrajan R, Narayanan N, Suba V, Bhaskar K. Simultaneous delivery of Nifedipine and Metoprololtartarate using sandwiched osmotic pump tablet system. Int J Pharm 2010;31:60-70.
- PrabakaranD,SinghP,KanaujiaP,MishraV,JaganathanKS,Vyas SP.Controlled porosity osmotic pumps of highly aqueous soluble drug containing hydrophilic polymers as release retardants. Pharm DevTechnol 2004;9:435-42.
- NokhodchiA,MominMN,ShokriJ,ShahsavariM,RashidiPA.Factors affecting the release of nifedipine from a swellable elementary osmotic pump.Drug Deliv 2008;15:43-8.
- ParkJS,ShinJH,LeeDH,KimMS,RheeJM,Lee HB,et al. A squeeze-type osmotic tablet for controlled delivery of nifedipine. BiomaterSciPolym 2008;19:31-45.
- WangCY,HoHO,LinLH,LinYK,SheuMT.Asymmetric membrane capsules for delivery of poorly water-soluble drugs by osmotic effects. Int J Pharm 2005;297:89-97.
- VenkataRatnam G, Ravi G, Harish G, Duraivel S, Pragati Kumar B. Formulation of venlafaxine sustained release capsule dosage form. J Chem Pharm Sci 2013;6:8-12.
- Gohel MC, SoniCD, Nagori SA,Sarvaiya KG. Modulation of venlafaxine hydrochloride release from press coated matrix tablet. Indian J Pharm Sci 2008;70:292-7.
- Omprakash B, Sav A, Gejage S, Amin P. Formulation development of venlafaxine hydrochloride extended release tablet andin vitrocharacterizations. Int J Pharm Tech Res 2012;4:1777-84.
- Sandhya P, Durani H, Majeed Z, Ganesh Kumar Y. Formulation and in vitroevaluation of sustained release tablets of venlafaxine hydrochloride by porous osmotic technology. Int J Sci Res Publ 2014;4:1-8.
- Singla D, Harikumar S. Osmotic pump drug delivery: a novel approach. Int J Res Pharm Chem 2012;2:661-70.
- Thanki K, Kulthe S, Mandge Y, Rao MRP. Formulation development and IVIVC of controlled porosity osmotic pump tablets of carvedilol phosphate. J Pharm Res 2011;4:4736-40.
- Wakode R, Bajaj A. Once a day osmotic drug delivery system for highly water soluble pramipexole. J Chem Pharm Res 2010;2:136-46.
- Verma R, Garg S. Formulation aspects in the development of osmotically controlled oral drug delivery system. J Control Release 2002;79:7-27.
- Ghedia T, Patel P, Christian V. Formulation and optimization of osmotic pump of propranolol hydrochloride using EOP and microporous concepts. Int J Res Pharm Sci 2011;2:590-5.
- Anusha N, Hindustan A, Naresh Babu G,Anilkumar K. Fabrication and in vitro evaluation of venlafaxine hydrochloride mini tablets filled hard gelatin capsules. Pharm Res 2013;1:161-8.
- Mangukia D, Patel C, Patel J. Formulation and evaluation of self-pore forming osmotic tablet of glipizide. Int Res J Pharm 2012;3:365-68.
- Indian Pharmacopoeia. Government of India, Ministry of Health and Family Welfare: Indian Pharmacopoeia Commission; 2014. p. 253-6.
- Sundaraganapathy R, Jambulingam M,Ananda T. Development and validation of UV spectrophotometric method for the determination of venlafaxine hydrochloride in bulk and solid dosage form. Int J Pharm Ind Res 2011;1:28-31.
- Verma RK, Garg S. Development and evaluation of osmotically controlled oral drug delivery system of glipizide. Eur J Pharm Biopharm 2004;57:513-25.
- Patel H, Patel MM. Formulation and evaluation of controlled porosity osmotic drug delivery system of carvedilol phosphate. J Pharm SciBioscientific Res 2012;2:77-82.
- Porsolt RD. Animal models of depression: utility for transgenic research. Rev Neurosci 2000;11:53-8.
- Petit-Demouliere B, Chenu F, Bourin M. Forced swimming test in mice. A review of antidepressant activity. Psychopharmacology 2005;177:245-55.
- Vogel HG. Drug discovery and evaluation. Pharmacological assays. 3rd ed. Germany: Springer-Verlag Berlin; 2008. p. 565-876.
- Chettapalli S, Renukadevi P, Gampa V. Stability indicating method of related impurities in venlafaxine hydrochloride sustained release tablets. Int J Adv Pharm Sci 2010;1:177-83.
- Dulin W. Oral Targeted Drug Delivery Systems: Enteric Coating. In: Wen H, Park K, editors. Oral Controlled Release Formulation Design and Drug Delivery: Theory to Practice. Hoboken (NJ): John Wiley and Sons Inc.; 2011. p. 205-19.
- Sinko PJ. Martin’s Physical Pharmacy and Pharmaceutical Sciences. 5th ed. New Delhi: Wolter Kluwer Health; 2010. p. 533-8.
- Gupta B, Thakur N, Jain N, Jain S. Osmotically controlled drug delivery system with associated drugs. J Pharm PharmSci 2010;13:571-88.
- Duarte A, Roy C, Gonzalez A, Duarte C. Preparation of acetazolamide composite microparticles by supercritical anti solvent techniques. Int J Pharm 2007;332:132-39.
- Giri T, Kumar K, Alexander A, Badwaik HA. Novel and alternative approach to controlled release drug delivery system based on solid dispersion technique. Bull Fac Pharm Cairo Univ 2012;50:147-59.
- Theeuwes F. Elementary osmotic pump. J Pharm Sci 1975;64:1987-91.
- http://www.chemtec.org/proddetail.php?prod=978-1-85957-479-9.
- Can A, Dao D, Chantelle E, Terrillion C, Piantadosi SC, Bhat S, et al. The tail suspension test. J Vis Exp 2012;59:3769.
- http://www.math.sci.hiroshima-u.ac.jp/stat/TR/TR06/TR06-07.pdf.