- *Corresponding Author:
- K. Kołodziejczyk
Department of Drug Form Technology, Chair of Applied Pharmacy, Medical University of Lodz, Poland, Muszynskiego 1, 90-151 Lodz, Poland
E-mail: michal.kolodziejczyk@umed.lodz.pl
| Date of Submission | 23 September 2018 |
| Date of Revision | 17 December 2018 |
| Date of Acceptance | 09 April 2019 |
| Indian J Pharm Sci 2019;81(3):489-495 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
The aim of the study was to analyze the effect of the active pharmaceutical ingredient on the flowability of the powder and properties of tablets prepared using direct compression method. Each analyzed formulation contained one model active pharmaceutical ingredient, diphenhydramine, ketoprofen or loratadine. Granulometric properties of the powder formulations were determined based on bulk density prior and after compression, Hausner ratio, Carr index and angle of repose values. The analysis of manufactured tablets included determination of their hardness, thickness, mass, disintegration time, wettability, contact angle and swelling index values. The preformulation study confirmed that characteristics of the active pharmaceutical ingredient might have modified physicochemical properties of both powder formulations and tablets.
Keywords
Orally disintegrating tablets, active pharmaceutical ingredient, flowability, wettability
Recent progresses in pharmaceutical sciences have been primarily oriented at optimization of the safety and efficacy of pharmacotherapy. Despite development of many innovative drug formulations, tablets are still most accepted by the patients, and direct compression technology is one of the most commonly used methods to produce solid drug forms for oral use [1-3]. Primary strengths of this method are its low cost and high throughput. However, aside from many advantages, this technique has some drawbacks too, such as the need for adequate flowability of tablet mass. The latter parameter is determined by numerous factors, such as the shape, size and surface of powder particles, their mutual interactions and environmental conditions such as humidity [4,5]. Low flowability of the powder may cause technological problems; the most serious of them include uneven filling of the tablet press, substantial differences in the mass and hardness of the tablets [3,6]. Moreover, one of the problems during drug production is the particle segregation. This means that there is no homogeneous distribution of the ingredients in the powder blend, and thus the uniformity of the manufactured tablets cannot be guaranteed. The main risk factor for segregation is the wide particle size distribution. In order to avoid this phenomenon, micronization of solid particles should be carried out. Micronization allows achievement of better physical properties of the drug such as changes in the morphological structure, increased powder mass homogeneity and the surface area of molecules in relation to the mass. The consequence of micronization is improvement of the dissolution rate of drug substances and bioavailability [7-10].
To satisfy the requirements of the compression process, the powder needs to have some specific characteristics. Also, tablets manufactured from the powder should have appropriate physical properties, such as hardness, wettability and disintegration time. The latter parameter is particularly important in case of tablets that need to dissolve quickly. Although orally disintegrating tablets (ODTs) have been known for more than 20 y, they continue to be popular, primarily because of a greater comfort of use compared to conventional tablets [11].
The properties of ODTs depend on many variables, including properties of excipients as well as the manufacturing process [12]. To obtain ODTs, various methods such as freeze drying, direct compression, granulation, spray drying, phase change and threedimensional printing can be used. However the direct compression technique distinguishes itself among other methods by simplicity and also it is the most desirable process in terms of time and economy [13]. Furthermore, in this type of process it is also possible to use moist or heat sensitive ingredients that would be contraindicated in the case of wet granulation [10]. Additionally, the choice of direct compression is rationalized from the fact that granulation can reduce superdisintegrants functionality in the meaning of water uptake and swelling ability [14].
Another factor that should be considered during development of tablet formulation is the nature of the active pharmaceutical ingredients (API). Differences in physicochemical properties of API can result in variation of properties of powder blends and tablets. Therefore, further studies concerning this correlation should be performed.
The aim of this study was to design a formulation for ODTs containing one of three model APIs, diphenhydramine or ketoprofen or loratadine. The APIs were the only element that differentiated the composition of the analyzed formulations. This enabled us to evaluate how the nature of an API influenced the flowability of the powder and properties of the tablets that were manufactured from it using direct compression method.
Materials and Methods
Ketoprofen (Medana Pharma Terpol Group S.A., s. 111.870), loratadine (Zydus Cadila, s. LT/002/3004), diphenhydramine hydrochloride (Beijing Taiyang Pharmaceutical Industry, s. M081105), microcrystalline cellulose, Vivapur 200 and Vivapur 105, and sodium starch glycolate, Vivastar P (JRS J. Rettenmaier & Söhne), sodium stearyl fumarate, PRUV (JRS Pharma), sodium lauryl sulphate (Sigma-Aldrich), methylene blue (POChSA Gliwice), were used in the present study. Korsch EKO single punch tablet press, TBH 125 hardness tester, TAR 220 abrasion tester and ZT 222 disintegration tester (Erweka GmbH), W-2 volumeter (ZDM Polfa), WTB 200 analytical scales (Radwag), CD-15CP caliper (Mitutoyo), sieve analyser (Haver and Boecker), Sieves, 1.0, 0.710, 0.5, 0.06, 0.045 and 0.032 mm, Petri dishes, object glasses, Eppendorf 20-200 μl automatic pipette, DeltaOptical microscope camera, Smart Analysis 1.0 software, were used.
Preparation of powder formulation:
Based on published data, 3 APIs with different physicochemical properties were chosen (Table 1). Diphenhydramine is an example of a hydrophilic compound. Loratadine is a hydrophobic drug, whereas ketoprofen exhibits indirect properties [15-18]. Moreover, diphenhydramine and loratadine are ideal candidates for incorporation into orodispersible tablets, because formulations for ODTs may exhibit rapid onset of action. This feature is important in case of definite medical conditions, such as episodes of allergic attack [19]. Selected model drugs were used to prepare three formulations for ODTs, differing solely in terms of their APIs (Table 2).
| Name of API | API 1 | API 2 | API 3 |
|---|---|---|---|
| Form according to FP XI | White or almost white crystalline powder | White or almost white crystalline powder | White or almost white crystalline powder |
| Solubility according to FP XI | Highly soluble in water, soluble in ethanol (96 %) | Virtually insoluble in water, soluble in acetone, ethanol (96 %) and methylene chloride | Virtually insoluble in water, soluble in acetone and methanol |
| BCS class | I | II | II |
| Log P | 3.27 | 3.12 | 5.20 |
| Melting point | 161-162° | 94° | 134-136° |
| Water solubility | 3.06 mg/ml | 0.051 mg/ml | 0.000011 mg/ml |
| Molecular weight | 291.8 g/mol | 254.3 g/mol | 382.9 g/mol |
| Chemical formula | C17H22ClNO | C16H14O3 | C22H23ClN2O2 |
| Pharmacological effect | Antihistaminic | Analgesic, Antiinflammatory, antipyretic |
Antihistaminic |
API 1 (active pharmaceutical ingredient 1) is diphenhydramine hydrochloride, API 2 is ketoprofen, API 3 is loratadine, BCS is Biopharmaceutics Classification System, FP XI - 11th edition of Polish Pharmacopeia
Table 1: Physicochemical properties of the active pharmaceutical ingredients
| Active ingredients/excipients | Amount (mg/ tablet) |
Formulation | ||
|---|---|---|---|---|
| F1 | F2 | F3 | ||
| Diphenhydramine | 18.75 | × | ||
| Ketoprofen | 18.75 | × | ||
| Loratadine | 18.75 | × | ||
| Microcrystalline cellulose type 105 | 72.19 | × | × | × |
| Microcrystalline cellulose type 200 | 51.19 | × | × | × |
| Sodium starch glycolate | 5.25 | × | × | × |
| Sodium stearyl fumarate | 1.31 | × | × | × |
| Sodium lauryl sulphate | 1.31 | × | × | × |
F1 is formulation 1 (with diphenhydramine hydrochloride), F2 is formulation 2 (with ketoprofen), F3 is formulation 3 (with loratadine)
Table 2: Ingredients of the analyzed tablets
After sieving through a sieve with a 0.5 mm mesh diameter, the ingredients for each formulation were weighed at an amount suitable for preparation of 400 tablets. The ingredients were micronized in a mortar. As a result, all components from formulation F1, F2 and F3 demonstrated the same size of particles. The obtained powder was sieved through a sieve with a 0.71 mm mesh diameter. The powder prepared as described above was subjected to flowability studies and used to manufacture tablets with direct compression method. Also, the tablets containing solely the excipients were prepared. Both types of tablets were pressed using spherical stamps with 8 mm in diameter. Furthermore, each of the three APIs were compressed into tablets using 11 mm spherical stamps.
Powder flowability studies, determination of angle of repose:
The analyses of powder masses included determination of the angle of repose, angle of slide, bulk density before and after the compression, Hausner ratio and Carr index values. To determine the angle of repose, a constant amount of the powder was poured freely through a glass funnel with a 10 mm outlet diameter on the surface of an immobile plate with a radius of 3.15 mm. During the test, the funnel was kept 2 cm above the top of the cone formed by the powder. Then, the height of the cone was measured and used to calculate the angle of repose from the following Eqn., tgα = h/0.5×d, where h is the height of the cone and d is the base diameter.
Determination of the angle of slide:
A constant amount of the powder was applied on one end of a 20×10 cm glass plate, and the opposite shorter edge of the plate was fixed on a flat surface. Then, the end of the plate with applied tablet mass was gradually elevated until the powder freely slid down the plate. The angle between the surface and the plate was recorded as the angle of slide.
Determination of bulk density before and after compression:
The test was conducted with an aid of a volumeter, in line with the 11th edition of Polish Pharmacopeia (FP XI) [18]. First, the volume of the powder and its weight were determined. Then, these parameters were used to calculate bulk density of the powder before and after compression from the following Eqns., ρ0 = powder mass/volume before compression×[g/ml], ρ1 = powder mass/volume after compression×(g/ml), where ρ0 is the bulk density before compression, ρ1 is the bulk density after compression. Based on the results, another two parameters, Hausner ratio and Carr index were calculated using the Eqns. presented below. Hausner ratio = ρ1/ρ0; Carr index = ρ1– ρ0/ρ1×100 (%). The same methodology was also used to determine the flowability of each of the APIs [18,20].
Determination of particle size distribution within the powder using sieve analysis:
The test was conducted on a sieve analyser, using 30 g of the powder and sieves with 0.5, 0.15, 0.071, 0.045 and 0.032 mm mesh diameters [18].
Determination of tablet properties:
Morphological assessment was done by determining the size and mass for 20 randomly selected tablets. Disintegration time was carried out as described in FP XI [18], with an aid of a tablet disintegration tester. The test was conducted on six randomly chosen tablets from each formulation, with distilled water with a temperature of 37° as an acceptor fluid. Crushability test was conducted with a hardness tester, on ten randomly chosen tablets, as described in FP XI [18]. Friability of non-coated tablets was conducted as described in FP XI [18], with an aid of an abrasion tester.
Water absorption index and wetting time:
Five millilitres of the solution containing a blue dye, methylene blue, dissolved in water was poured to a 5 cm Petri dish. Then, five rings of filtration paper were placed in the dish, one on another. A previously weighed tablet was applied onto the surface of the paper rings, and its wetting time was measured until the whole upper surface of the tablet was colored in blue. Then, the tablet was weighed again. The test was conducted for ten tablets from formulations F1, F2 and F3, as well as for tablets that contained solely the excipients. The results were used to calculate water absorption index, using the following Eqn., % R = Wa– Wb/Wb×100, where Wa is tablet weight after wetting, Wb is tablet weight before wetting [21].
Wettability was determined based on the value of contact angle measured with a sessile drop method. The contact angle is formed by the tangent of the droplet applied onto the examined tablet and flat surface of the latter [18]. To determine the contact angle, the tablet was placed on a flat surface, and a single drop of water was applied with a micropipette. Immediately after that, an image was taken with an aid of a microscope camera and used to estimate the contact angle with Smart Analysis software. The measurement was repeated seven times for each formulation.
Minimum volume of water needed for complete wetting of the tablet was determined after placing the tablet on a Petri dish, a prespecified volume of water was applied onto its surface, and the degree of tablet wetting was estimated by palpation. First, 0.5 ml of water was applied, which corresponded to a baseline secretion of saliva in human oral cavity [22]. Subsequently, the amount of applied water was reduced at 0.1 ml increments down to 20 μl, and then, at 5 μl increments. The test was repeated three times.
True density was calculated using the following Eqn., Dt = 4mt/πhd2, where Dt is true density (g/cm3), mt is an average mass of tablet (g), h is an average thickness of tablet (cm) d is an average diameter of tablet (cm) [23].
Results and Discussion
The flowability of three powder mixtures, formulations for ODTs, containing APIs with different physicochemical properties was analysed. Furthermore, the properties of tablets manufactured from the powders using direct compression method was determined using both pharmacopoeial and nonpharmacopoeial methodology [21].
The sieve analysis demonstrated that irrespective of analyzed formulation, the largest portion of the powder was retained on a sieve with a 0.15 mm mesh size. For F2 and F3 formulations, the distribution of particles on the remaining sieves was even, whereas the distribution of particles within the F1 formulation followed the pattern: 0.15>0.032>0.071>0.045 mm (Table 3).
| Mesh diameter (mm) | Total mass of powder applied on the sieve (g) | Proportion of initial mass retained on the sieve (%) | ||||
|---|---|---|---|---|---|---|
| F1 | F2 | F3 | F1 | F2 | F3 | |
| 0.15 | 10.569 | 8.032 | 8.126 | 35.21 | 26.77 | 27.01 |
| 0.071 | 3.827 | 3.962 | 4.306 | 12.75 | 13.2 | 14.31 |
| 0.045 | 2.001 | 7.332 | 7.061 | 6.67 | 24.44 | 23.47 |
| 0.032 | 9.161 | 6.436 | 6.741 | 30.52 | 21.45 | 22.41 |
F1 is formulation 1 (with diphenhydramine hydrochloride), F2 is formulation 2 (with ketoprofen), F3 is formulation 3 (with loratadine)
Table 3: Sieve analysis of formulations F1, F2 and F3
The study demonstrated that powder formulation F1 was the most flowable, as shown by the lowest values of the angle of repose and the angle of slide. In line with the criteria published in FP XI, the flowability of this formulation was relatively good. Formulations F2 and F3 had worse flowability, merely sufficient according to the pharmacopoeial standards [18]. Formulation F1 had the highest bulk density, both before and after the compression. No substantial between-formulation differences were documented in Hausner ratio and Carr index (Table 4).
| Formulation/ API | Repose angle | Flowability (FP XI) |
Bulk density (g/ml) | Hausner ratio | Carr index | Slide angle | |
|---|---|---|---|---|---|---|---|
| before | after | ||||||
| API 1 | 33.00° | good | 0.4838 | 0.5760 | 1.1906 | 16.01 % | 37.60° |
| API 2 | 51.00° | poor | 0.3667 | 0.5238 | 1.4284 | 29.99 % | 44.50° |
| API 3 | 49.00° | poor | 0.2181 | 0.2948 | 1.3517 | 26.02 % | 34.40° |
| F1 | 39.67° | relatively good | 0.4306 | 0.5820 | 1.3516 | 26.01 % | 39.33° |
| F2 | 41.33° | sufficient | 0.3552 | 0.4800 | 1.3514 | 26.00 % | 49.30° |
| F3 | 43.00° | sufficient | 0.3894 | 0.5192 | 1.3333 | 25.00 % | 47.33° |
F1 is formulation 1 (with diphenhydramine hydrochloride), F2 is formulation 2 (with ketoprofen), F3 is formulation 3 (with loratadine), API 1 (active pharmaceutical ingredient 1) is diphenhydramine hydrochloride, API 2 is ketoprofen, API 3 is loratadine
Table 4: Granulometric parameters of analyzed formulations and active pharmaceutical ingredients
A significant correlation was found between powder and API flowability. Both diphenhydramine and formulation F1 containing this API were characterized by the best flowability, whereas the flowability of the other two active ingredients and respective formulations, F2 and F3, were worse, which might correspond to their greater cohesiveness resulting from stronger interparticle forces.
Three formulations of ODTs were manufactured using direct compression. The tablets were white, with smooth surface free from visible abrasions. Physicochemical characteristics of the tablets are presented in Table 5.
| Parameter (Units) | Formulation | ||
|---|---|---|---|
| F1 | F2 | F3 | |
| Mass (mg) | 148.905±2.500 | 145.800±3.563 | 157.000±3.680 |
| Diameter (mm) | 9.029±0.004 | 9.021±0.003 | 9.021±0.002 |
| Thickness (mm) | 2.459±0.013 | 2.497±0.009 | 2.469±0.014 |
| Disintegration time (s) | 15.5 | 7.0 | 21.5 |
| Friability (%) | 0.21 | 0.26 | 0.18 |
| Hardness (N) | 56.90±14.27 | 45.45±2.01 | 67.65±10.78 |
| Vmin of water for complete wetting (µl) | 80 | 90 | 85 |
| Water absorption index (%) | 132.135±27.546 | 234.259±13.123 | 260.005±16.342 |
| Wetting time (s) | 22.000±1.563 | 16.000±0.943 | 24.875±2.997 |
| True density (g/cm3) | 0.9462 | 0.9140 | 0.9954 |
F1 is formulation 1 (with diphenhydramine hydrochloride), F2 is formulation 2 (with ketoprofen), F3 is formulation 3 (with loratadine), Vmin is minimum volume of water needed for complete wetting of tablets (µl)
Table 5: Physicochemical characteristics of tablets prepared from formulations F1, F2 and F3
| Tablet | Contact angle | |||
|---|---|---|---|---|
| mean | minimum | maximum | standard deviation | |
| F1 | 18.38° | 18° | 19° | 0.47 |
| API 1 | 13.70° | 12° | 16° | 1.70 |
| F2 | 21.80° | 19° | 25° | 1.77 |
| API 2 | 53.50° | 52° | 56° | 1.38 |
| F3 | 34.80° | 34° | 36° | 0.90 |
| API 3 | 56.00° | 54° | 57° | 1.41 |
| Excipients | 18.00° | 16° | 20° | 1.49 |
F1 is formulation 1 (with diphenhydramine hydrochloride), F2 is formulation 2 (with ketoprofen), F3 is formulation 3 (with loratadine), API 1 (active pharmaceutical ingredient 1) is diphenhydramine hydrochloride, API 2 is ketoprofen, API 3 is loratadine, standard deviation for n=7 observations
Table 6: Contact angles for analyzed formulations
Standard deviation from mean mass of each tablet formulation remained within the pharmacopoeial limit [18]. The lowest value of this parameter was determined for formulation F1, which might result from its better flowability and lesser contribution of interparticle cohesive forces.
Each formulation showed adequate friability, consistent with pharmacopoeial standards [18]. Formulation F3 had greater mechanical resistance than formulations F1 and F2, as shown by its greatest hardness and lowest friability.
The tablets from all formulations satisfied the pharmacopoeial standards for disintegration time (less than 3 min) [18]. Moreover, the disintegration time for all tablets was consistent with the US Food and Drug Administration requirements according to which, this parameter should not be longer than 30 s [24]. Formulation F3 had the longest disintegration time (19 s) of all the analyzed tablets.
The minimum volume of water (Vmin) needed for complete wetting of the tablets from each formulation was smaller than the baseline secretion of human saliva. Analysed formulations differed in terms of their swelling indices and wetting times (Table 5). The highest values of both parameters were observed for formulation F3. Formulation F1 was characterized by the lowest value of water absorption index, and formulation F2 had the shortest wetting time. The highest volume of water was absorbed by the tablets manufactured solely from the excipients (water absorption index 317.366 %); the addition of API worsened the swelling indices of the tablets. Moreover, regardless of the formulation, the addition of API contributed to a substantial increase in wetting time, which was substantially longer than in the case of the tables without any APIs (4.2 s).
A significant linear correlation was found between the wetting time and tablet hardness. Moreover, the wetting time correlated significantly with tablet disintegration time (fig. 1). Hence, greater density of the tablet resulted in longer wetting time, which was in turn reflected by longer disintegration time. The lowest value of contact angle implied that formulation F1 was characterized by the best wettability of tablet surface. Formulation F3 turned out to be the most hydrophobic, as shown by the highest value of the contact angle. Contact angle values for the tablets composed solely of the APIs followed the analogous pattern, i.e. API1<API2<API3 (Table 6, fig. 2). The addition of API to the formulation contributed to an increase in contact angle value, the most evident for formulation F3.

Figure 1: Relationship of wetting time with hardness and
disintegration time
Relationship of wetting time with (a) hardness and (b)
disintegration time of the tablets from 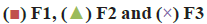

Figure 2: Contact angles for formulations
(a) F1 with the mean contact angle of 18.38°, (b) F2 with the mean contact angle of 21.80° and (c) F3 with the mean contact angle
of 34.80°
In conclusion, characteristics of API seem to influence the flowability of powder formulations. In this study, the best flowability was observed for the formulation with diphenhydramine, as shown by both granulometric properties of the powder formulation and standard deviation from the mass of manufactured tablets. Hence, a change of API within the formulation seems to contribute to differences in physicochemical properties of manufactured tablets. All analyzed formulations satisfied the pharmacopoeial criteria for tablets, which implies that they could be used on a larger scale than in a laboratory setting. Disintegration time of the tablet may be estimated based on its wetting time. Lower hydrophilicity of the API may contribute to better swelling ability of the tablet, which results from hydrophobic interactions creating spaces for water penetrating into the tablet. Greater lipophilicity of the API (higher value of octanol/water separation index) may be a factor contributing to longer disintegration time of the tablet. Weaker hydrophilic properties of the API may be reflected by greater hydrophobicity of the tablet’s surface, which in turn contributes to its lesser wettability. In addition, it is worth noting that many factors during manufacturing process, such as mechanical stress, compaction pressure or temperature increase may induce phase transformation of the API. This modification can influence the properties of the final dosage form like wettability of tablet’s surface.
Financial interest and scholarship:
This research was financially supported by the grant (503/3-021-02/503-31-002) from Medical University of Lodz, Poland.
References
- Preis M, Breitkreutz J, Sandler N. Perspective: Concepts of printing technologies for oral film formulations. Int J Pharm2015;494:578-84.
- Zhang J, Yang W, Vo AQ, Feng X, Ye X, Kim DW, et al. Hydroxypropyl methylcellulose-based controlled release dosage by melt extrusion and 3D printing: Structure and drug release correlation. Carbohydr Polym2017;177:49-57.
- Sznitowska M. Farmacja stosowana technologia postaci leku. Warszawa: Wydawnictwo Lekarskie PZWL; 2017. p. 3-6, 253-274, 281.
- Capece M, Silva KR, Sunkara D, Strong J, Gao P. On the relationship of inter-particle cohesiveness and bulk powder behavior: Flowability of pharmaceutical powders. Int J Pharm 2016;511:178-89.
- Juarez-Enriquez E, Olivas GI, Zamudio-Flores PB, Oretga-Rivas E, Perez‑Vega S, Sepulveda DR. Effect of water content on the flowability of hygroscopic powders. J Food Eng2017;205:12-7.
- Nachajski M. Proszkinie tylko recepturowe. [cited 2017 Dec 7]. Available from: http://www.aptekarzpolski.pl/2012/04/04-2012-proszki-nie-tylko-recepturowe.
- Jachowicz R, Leku P. Optymalizacja leków doustnych i do oczu w nowoczesnej technologii farmaceutycznej. Warszawa: Wydawnictwo Lekarskie PZWL; 2013, p. 141-3.
- Khadka P, Ro J, Kim H, Kim J, Kim JT, Kim H, et al. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J Pharm Sci 2014;9(6):304-16.
- Martínez-Acevedoa L, Zambrano-Zaragoza ML, Vidal-Romero G, Mendoza-Elvira S, Quintanar-Guerrero D. Evaluation of the lubricating effect of magnesium stearate and glyceryl behenate solid lipid nanoparticles in a direct compression process. Int J Pharm 2018;545(1-2):170-5.
- Meeus L. Direct Compression versus Granulation. [cited 2018 Dec 3]. Available from: http://www.pharmtech.com/direct-compression-versus-granulation.
- Montgomery W, Treuer T, Karagianis J, Ascher-Svanum H, Harrison G. Orally disintegrating olanzapine review: effectiveness, patient preference, adherence, and other properties. Patient Prefer Adherence 2012;6:109-25.
- Gandhi A. Mouth Dissolving Tablets: A New Venture in Modern Formulation Technology. Pharm Innov 2012;1(8):14-31.
- Gulsun T, Cayli YA, Izat N, Cetin M, Oner L, Sahin S. Development and evaluation of terbutaline sulfate orally disintegrating tablets by direct compression and freeze drying methods. J Drug Deliv Sci Tec 2018,46:251-8.
- Zhao N, Augsburger LL. The Influence of Granulation on Super Disintegrant Performance. Pharm Dev Technol 2006;11(1):47-53.
- Diphenhydramine hydrochloride, DrugBank. [cited 2017 Dec 7]. Available from: https://www.drugbank.ca/salts/DBSALT000056.
- Ketoprofen, DrugBank. [cited 2017 Dec 7]. Available from: https://www.drugbank.ca/drugs/DB01009.
- Loratadine, DrugBank. [cited 2017 Dec 7]. Available from: https://www.drugbank.ca/drugs/DB00455.
- Farmakopea PolskaXI. 11th ed. Warszawa: Polskie Towarzystwo Farmaceutyczne; 2017.
- Kumar E, Bhagyashree J. Mouth Dissolving Tablets – A comprehensive Review. Int J Pharm Res Rev 2013;2(7):25-41.
- Skoczeń P, Gąsior P, Sawicki W. Znaczenie odpowiedniej zsypywalności masy tabletkowej w procesie kompresji. Farm Pol2011;67:859-66.
- Battu SK, Repka MA, Majumdar S, Madhusudan RY. Formulation and evaluation of rapidly disintegrating fenoverine tablets: Effect of superdisintegrants. Drug Dev Ind Pharm2007;33:1225-32.
- Cyprysiak G, Tadeusiak W. Zastosowanie śliny w diagnostyce medycznej. [cited 2018 Feb 19]. Available from: http://www.czytelniamedyczna.pl/1975,zastosowanie-sliny-w-diagnostyce-medycznej.html.
- Linka WA, Wojtaszek I, Zgoda MM, Kołodziejczyk MK. The Use of Synthetic Polymers (Superdisintegrants) in Technology Tablets Containing Ethanol Dry Extract from Asparagus officinalis. Polim Med 2015;45(2):73-80.
- Food and Drug Administration, Draft Guidance for Industry, Orally Disintegrating Tablets. [cited 2017 Dec 21]. Available from: https://www.fda.gov/downloads/Drugs/.../Guidances/ucm070578.pdf.