- *Corresponding Author:
- S. Y. Rai
SVKM’s Dr. Bhanuben Nanavati College of Pharmacy, Gate No. 1, V. M. Road, Vile Parle (W), Mumbai-400056, India
E-mail: sanjurai2011@gmail.com
| Date of Submission | 18-Apr-2015 |
| Date of Revision | 01-Feb-2016 |
| Date of Acceptance | 19-Mar-2016 |
| Indian J Pharm Sci 2016;78(2):182-192 |
Abstract
The aim of this work is to develop microsphere formulation and study the impact of drug-polymer ratio, surfactant concentration and stirring speed on particle size and drug entrapment during preparation. Clotrimazole loaded Eudragit S100 microspheres were prepared by emulsion solvent evaporation method. A three level factorial Box Behnken design was used to characterize and optimize the formulation. The result of analysis of variance test indicated that the test is significant. The optimum drug: polymer ratio (X1), surfactant concentration (X2) and stirring speed (X3) was found to be 1:4, 1.5% and 1000 rpm respectively to obtain particle size and maximum entrapment efficiency of microspheres as 35.6 µm and 83.3%, respectively. The drug release from microsphere based cream exhibited a controlled release pattern.
Keywords
Microspheres, emulsion solvent evaporation, controlled release, Box-Behnken design.
Several drugs are employed topically to treat skin fungal infections. Azoles are most prominent therapeutic agents among them. Clotrimazole is an imidazole derivative with a broad spectrum of antimycotic activity. It inhibits biosynthesis of the sterol ergostol, an important component of fungal cell, and thus results in antifungal activity. Topically applied antifungal azoles sometimes cause skin irritation/systemic absorption which lead to the therapeutic failure [1]. To overcome such adverse effects of antifungal drugs, use of novel drug delivery systems have been proposed for the treatment of skin fungal infections. Several novel drug delivery systems like liposomes, niosomes, ethosomes, microemulsions, nanoparticles, micelles and microspheres were explored for topical antifungal therapy [2].
Microparticles designed to control drug release into the skin ensure that the drug remains localized at the application site and does not unnecessarily enter into the systemic circulation [3]. They act as reservoir releasing an active ingredient over an extended period of time maintaining effective drug concentration in the skin and at the same time, reducing undesired side effects [4]. Thus, periods of over-medication and under-medication are eliminated. This is especially important in the treatment of infectious diseases due to the prevention of antimicrobial resistance. Such delivery systems may also increase drug stability or improve incorporation into appropriate vehicles [5].
In the present work, clotrimazole microspheres were prepared by emulsion solvent evaporation technique using Eudragit S100 as the microencapsulating polymer. Factorial design was applied to study the effect of formulation and process variables. The drug:polymer ratio, surfactant concentration and stirring speed were selected as independent variables while the particle size and entrapment efficiency were chosen as the dependant variables.
Materials and Methods
Clotrimazole BP was a gift sample from Ciron Drugs Pvt Ltd., Palghar. Eudragit S100 was received as gift sample from Evonik Industries, Mumbai. Sodiumcarboxy methylcellulose was obtained from Signet Chemical Corporation Pvt Ltd., Mumbai. Fungal strain of Candida albicans culture was obtained from Mithibai College, Mumbai. All other solvents, reagents and chemicals used were of analytical (AR) grade and purchased from SD Fine- Chem Ltd., Mumbai.
Preparation of microspheres
Microsphere formulations using Eudragit S100 as a carrier polymer were prepared using emulsion solvent evaporation technique. Desired quantity of Eudragit S100 polymer was dissolved in 10 ml of chloroform to form a homogenous polymer solution. Calculated quantity of drug was added to this polymer solution and mixed thoroughly. The resulting mixture was then added to 250 ml of aqueous mucilage of sodium CMC (0.5%) containing 1% v/v tween 80, while stirring at 1000 rpm for emulsification. Chloroform was removed by evaporation during continuous stirring at room temperature for 3 h to produce spherical microspheres. Chloroform was used as the polymer solvent, aqueous mucilage of sodium CMC as microencapsulating vehicle and tween 80 as dispersing agent. During 3 h of stirring period, chloroform was completely removed by evaporation. Microspheres were collected by vacuum filtration, washed repeatedly with distilled water and petroleum ether and dried at room temperature for 24 h to get free flowing microspheres [6].
Preparation of microspheres loaded cream
Oil phase containing stearic acid, isopropyl myristate and aqueous phase containing propylene glycol, triethanolamine and water were weighed in separate beakers and heated to 65º in a water bath. After reaching 65º, aqueous phase was added to the oil phase and stirred to form a homogenous mixture. Mixture was cooled at room temperature to get smooth, white cream. The formulated microspheres were then uniformly dispersed in the cream base by mechanical stirring for 30 min to get microspheres loaded cream. Conventional cream containing 1% w/w clotrimazole was also prepared in the same manner.
Experimental design for optimization
Clotrimazole microspheres were prepared and evaluated using “Box-Behnken experimental design”. Aim of this study is to statistically optimize the formulation parameters of clotrimazole loaded microspheres for maximum entrapment and optimum particle size. Variables selected were polymer amount (X1), surfactant concentration (X2) and stirring speed (X3). The response variables were mean particle size (Y1) and entrapment efficiency (Y2) of microspheres. The levels for these variables were determined from the preliminary trials and are shown in Table 1.
| Independent variables | Levels used, actual(coded) | ||
|---|---|---|---|
| Low (-1) | Medium (0) | High (1) | |
| A= Drug: polymer ratio | 1:1 | 1:2 | 1:4 |
| B= Surfactant concentration (%) | 0.5 | 1 | 1.5 |
| C= Stirring speed (rpm) | 800 | 1000 | 1200 |
Table 1: Optimization Levels
Box-Behnken design was used to statistically optimize the formulation factors and evaluate effects such as main, interaction and quadratic; on the entrapment efficiency and particle size. A three factor, threelevel Box-Behnken statistical experimental design as the Response Surface Methodology requires 17 runs and these 17 runs with triplicate centre points were generated. The full factorial design and layout with coded values of variables for each batch and responses are shown in Table 2. The ranges of Y1 and Y2 for all batches were 25 to 50 µm and 55-84%, respectively. All the responses observed for the 17 formulations were simultaneously fitted to various models like first order, second order and quadratic, using Design Expert® (Version 9, Stat-Ease Inc.). It was observed that the best-fitted model was quadratic and the comparative values of R2, Standard deviation and % CV are given in Table 3.
| Runs | Factor A Drug: polymer ratio |
Factor B Surfactant concentration (%) |
Factor C Stirring speed (rpm) |
Response 1 Particle size (µ) |
Response 2 Entrapment Efficiency (%) |
|---|---|---|---|---|---|
| 1 | -1 | -1 | 0 | 35.29 | 73.2 |
| 2 | 1 | 1 | 0 | 35.6 | 83.3 |
| 3 | 0 | 1 | -1 | 40.69 | 61.9 |
| 4 | 0 | -1 | 1 | 29.49 | 55.3 |
| 5 | -1 | 1 | 0 | 33.63 | 66.1 |
| 6 | 0 | 1 | 1 | 25.25 | 65.2 |
| 7 | 1 | -1 | 0 | 48.66 | 79.9 |
| 8 | 1 | 0 | -1 | 41.87 | 77.8 |
| 9 | 0 | 0 | 0 | 32.89 | 75.1 |
| 10 | 0 | -1 | -1 | 38.24 | 72.1 |
| 11 | 1 | 0 | 1 | 34.68 | 66.4 |
| 12 | 0 | 0 | 0 | 30.52 | 77.6 |
| 13 | 0 | 0 | 0 | 29.37 | 69.9 |
| 14 | 0 | 0 | 0 | 30.24 | 69.8 |
| 15 | -1 | 0 | -1 | 33.66 | 56.6 |
| 16 | 0 | 0 | 0 | 28.86 | 72.3 |
| 17 | -1 | 0 | 1 | 25.12 | 58.1 |
Table 2: Design Summary
| Response | R-Squared | Adj R-Squared |
Pred R-Squared |
Adeq. Precision | Std. dev | CV % | Model | |
|---|---|---|---|---|---|---|---|---|
| F value | P-value >F | |||||||
| Particle Size | 0.9323 | 0.8453 | 0.1518 | 12.495 | 2.4 | 7.1 | 10.71 | 0.0025 |
| Entrapment Efficiency | 0.9514 | 0.8890 | 0.8314 | 13.402 | 2.75 | 3.96 | 15.23 | 0.0008 |
Table 3: Statistical Analysis
Two dimensional contour plots were prepared for both the responses and are shown in figs. 1 and 3 for responses Y1 and Y2, respectively. The threedimensional response surface plots are shown in figs. 2 and 4 for responses Y1 and Y2, respectively. These plots are known to study the interaction effects of the factors on the responses as well as are useful in studying the effects of two factors on the response at one time.

Fig. 1: Two dimensional contour plots for the effect of drug, polymer ratio and surfactant concentration on particle size.
a. Effect of drug:polymer ratio and surfactant concentration; b. effect of drug:polymer ratio and stirring speed; c. effect of surfactant concentration and stirring speed

Fig. 2: Three dimensional contour plots for the effect of drug, polymer ratio and surfactant concentration on particle size.
a. Effect of drug:polymer ratio and surfactant concentration; b. effect of drug:polymer ratio and stirring speed; c. effect of surfactant concentration and stirring speed

Fig. 3: Two dimensional contour plots for the effect of drug, polymer ratio and surfactant concentration on entrapment efficiency.
a. effect of drug:polymer ratio and stirring speed; b. effect of surfactant concentration and stirring rate; c effect of drug:polymer ratio and surfactant concentration

Fig. 4: Three dimensional contour plots for the effect of drug, polymer ratio and surfactant concentration on entrapment efficiency.
a. effect of drug:polymer ratio and surfactant concentration; b. effect of drug:polymer ratio and stirring speed; c. effect of surfactant concentration and stirring rate
Response 1- Particle size (Y1)
The model proposes following polynomial equation for particle size, Particle size
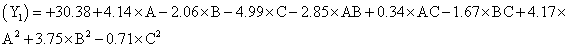
Where, Y1 is the particle size in µ, A is polymer amount, B is surfactant concentration and C is the stirring rate. The model F-value of 10.71 implies the model is significant (P<0.0025). The lack of fit F-value of 4.22 implies the lack of fit is not significant (P=0.0991). The predicted R squared of 0.1518 is not in reasonable agreement with the adjusted R-squared of 0.8453. Signal to noise ratio greater than 4 is desirable. Our ratio of 12.495 indicates an adequate signal and hence this model can be used to navigate the design space.
Response 2- Entrapment efficiency (Y2)
The following polynomial equation was proposed from the model for entrapment of clotrimazole in the microspheres. Entrapment efficiency
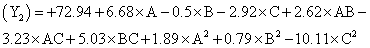
Where, Y2 is entrapment efficiency. The model was found to be significant (F-value=15.23; P=0.0008). In this case A, C, BC, C2 are significant model terms. The predicted R-squared is in reasonable agreement with the adjusted R-squared (0.8314 and 0.8890, respectively). Signal to noise ratio greater than 4 is desirable. Our ratio of 13.402 indicates an adequate signal.
All statistically significant (P<0.05) coefficients are included in the equations in which positive value shows an effect that favours the optimization, while a negative value represents an inverse relationship between on the response. Upon trading of various response variables, the formulation composition of batch number 2 was found to fulfil the maximum requisite of an optimum formulation because of maximum entrapment and optimum particle size.
Formulation optimization
A numerical optimization technique using the desirability approach was employed to develop an optimized formulation with the desired responses. For the optimization of clotrimazole microspheres, constraints were fixed for all factors and responses. In optimization, maximum desirability of 0.759 indicated an optimum formulation which was achieved at polymer amount (1), surfactant concentration (1) and stirring speed of (0), which was nearest to the batch 2. Overlay plot (fig. 5) of the desirability gives the details of the optimized batch producing best results. Thus, it can be concluded that batch 2 may be considered as the optimized batch.

Fig 5: Overlay plot.
Drug polymer interaction (FTIR) study
FTIR spectroscopy was performed on Fourier transform infrared spectrophotometer. The drug and potassium bromide were mixed and pellets were prepared by compressing the powders at 100 kg/cm2 for 1 min on KBr-press and the spectra were scanned in the wave number range of 4000-400 cm-1. FTIR study was carried out on drug, physical mixture of drug and polymer, drug loaded microspheres.
Surface morphology (SEM)
Scanning electron microscopy has been used to determine particle size, surface morphology and texture. SEM studies were carried out by using scanning microscope. Dry microspheres were placed on an electron microscope stub covered with a black adhesive tape and observed under microscope after applying vacuum. Picture of microspheres were taken by random scanning of the stub.
Size distribution analysis
Optical microscopy was used to carry out determination of average particle size of microspheres. A minute quantity of microspheres was spread on a clean glass slide and average size of 50 microspheres was determined in each batch.
Percentage yield
Percentage practical yield is calculated to know about efficiency of the method of preparation. Practical yield is calculated as the weight of microspheres recovered from each batch in relation to the sum of starting material. The percentage yield was determined by using the formula, % yield=(actual weight of the product/total weight of polymer and drug)×100.
Entrapment efficiency
Efficiency of drug entrapment in the microspheres for each batch was calculated in terms of percentage drug entrapment as per the following formula [7], % entrapment efficiency= (practical drug content/ theoretical drug content)×100.
For this, 10 mg of drug containing microspheres were dissolved in 10 ml methanol and drug content was analysed by UV spectroscopy against blank methanol solution containing 10 mg of blank microspheres.
Differential Scanning Calorimetry (DSC)
The physical state of clotrimazole in the microspheres was analyzed by Differential Scanning Calorimeter. The thermograms of the drug, polymer and drug loaded microspheres were obtained at a scanning rate of 10°/min conducted over a temperature range of 25–300°.
Evaluation of microsphere based cream
All the creams were tested for homogeneity by visual inspection. pH of formulation was determined by dispersing 0.5 g of cream in 5 ml of water by using digital pH meter at constant temperature. Prior to this, pH meter was calibrated using buffer solutions of pH 4, 7, 10 and the electrode was washed with demineralised water.
Weighed amount of 1 g of the cream was dissolved in 100ml of pH 6.8 phosphate buffer and stirred for 24 h. The solution was diluted to appropriate concentration. The absorbance was taken at 258.6 nm and the concentration of drug was calculated by using the equation of standard graph.
Spreadability is a property of topical formulation from a patient compliance point of view. An excess of cream sample (1 g) was placed between two glass slides and a 500 g weight was placed on upper slide for few minutes to compress and uniformly spread the cream between the slides. A specific weight of 10 mg was placed on the pan and increased slowly till the slide started sliding. The time required to separate the two slides was taken as a measure of spreadability and it was calculated using the formula, S=M×L/T.
Where, S=spreadability (g.cm/s), L=length of the glass slide (cm), T=time (s), M=weight tied to the upper slide (g) [8].
Viscosity measures the flow characteristics of topical formulation. Changes in viscosity of the product are indicative of changes in stability or effectiveness of the product. Viscosity was determined by using spindle number 4 at 50 rpm.
In vitro Permeation Studies
Franz-diffusion cell with receptor volume 22 ml and diffusional area of 3.14 cm2 was used. Dialysis membrane 150 was used as a diffusion membrane and was placed between donor and receptor compartments of the Franz-diffusion cell. The receptor chamber was filled with fresh phosphate buffer, pH 6.8 and 1 g microsphere cream was loaded in the donor compartment. Aliquots (2 ml) of receptor medium were withdrawn and replaced with fresh medium at specified intervals of time i.e. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10,12 and 24 h and analyzed by UV.
Ex vivo Permeation Studies
Ex vivo permeation study of clotrimazole from microsphere cream was investigated using porcine ear skin. Extraneous subcutaneous fat was removed from the surface of the porcine ear and it was stored in a freezer at -20° until utilized. Skin was slowly thawed and was cut into small circular pieces of approximately 2.5 cm diameter. The skin was allowed to hydrate for 1 h at 37° in the receptor medium prior to experimentation. The porcine skin was mounted carefully onto the diffusion cell with the stratum corneum side facing the donor compartment. After clamping the donor and receiver compartments together, the receiver compartment was filled with phosphate buffer, pH 6.8 and stirred with a magnetic stirrer. At different time intervals, the aliquots from receptor solution were withdrawn and replaced with equal volumes of fresh phosphate buffer, pH 6.8 maintained at 37°. Each experiment was performed in triplicate.
Kinetic analysis
The data obtained from the release studies were kinetically analyzed to determine the mechanism and the order of drug release from various formulations. Linear regression analysis was done to fit the data to various models like zero order, first order, Higuchi and Korsemeyer-Peppas [3].
Stability studies
Samples were kept at 8°±2°, 25°±2°/60±5% RH, 40°±2°/75±5 % RH [9] for three months. Samples were withdrawn periodically at time intervals of 1st, 2nd and 3rd month and evaluated for parameters such as pH, viscosity, drug content, homogeneity and phase separation. Stability data for each attribute was assessed sequentially as per ICH guidelines.
Skin irritation test
The protocol was approved by SVKM’s Dr. Bhanuben Nanavati College of Pharmacy’s Institutional Animal Ethical Committee (IAEC) bearing approval no. CPCSEA/IAEC/BNCP/P-09/2014.Six healthy male Albino Wistar rats (Rattus norvegicus), weighing 180-200g were used for the study. All the animals were allowed to acclimatize to laboratory conditions for a period of 48 h prior to study. Hair was removed from the back side of rats and an area of 1 cm2 was marked on both the sides of which one side served as control while the other as test. Control (conventional marketed cream) and test formulation (Microsphere based cream, 200 mg/rat) were applied on each side and the site was covered with cotton bandage. Observations for sensitivity and reaction were made after 24, 48 and 72 h and graded as; Grade 1: No reaction; Grade 2: Slight, patchy erythema; Grade 3: Slight but confluent or moderate, well defined erythema; Grade 4: Moderate to severe erythema; Grade 5: Severe erythema with/without edema [10].
Antifungal activity
Antifungal activity of the final formulation was checked by cup-plate method. A definite volume of the Candida albicans suspension (innoculum) was poured into the sterilized dextrose agar media, cooled at 40° and mixed thoroughly. About 20 ml of this medium was poured aseptically in the petriplates and kept to solidify. The surface of the agar plates was pierced using a sterile cork borer. Standard solution of clotrimazole, optimized batch of microsphere cream and conventional cream having same strength were filled in the prepared wells. After that it was incubated at 28° for 24 h. The fungal growth was observed and the zone of inhibition was measured using antibiotic zone reader [11].
Results and Discussion
Preparation of clotrimazole loaded microspheres was carried out by emulsion solvent evaporation method. Various process and formulation variables employed during the studies like stirring speed, concentration of surfactant and drug: polymer ratio were evaluated for optimization. Based on the results of the particle size and entrapment efficiency, Batch 2 was selected as optimized batch and further converted into topical cream. Drug polymer interaction was studied by FTIR spectroscopy. No interaction between drug and polymer was seen which can be interpreted from the fig. 6 as there is no major shift in the peaks of drug spectra. FTIR spectroscopy of microsphere is given in fig. 6c. Scanning electron microscopy (fig. 7) showed that the microspheres were spherical in shape and surface was free from drug crystals. Particle size of microspheres was found in the range of 25-50 µm. Percent yield of all the batches were in the range of 65 to 90% and it was highest for batch number 2. Entrapment efficiency of different trials was found to be in the range of 58-83% and maximum entrapment of 83.3% was seen in case of batch 2 where drug: polymer ratio was 1:4, surfactant concentration 1.5 % and stirring speed 1000 rpm. High entrapment of clotrimazole may be due to its water insolubility resulting in accumulation of drug in organic phase during emulsion formation due to preferred partitioning. DSC spectra of pure drug and microspheres are shown in figs. 8a and 8b, respectively. Pure clotrimazole showed a sharp endothermic peak of melting at about 147.9°, but no related peak was observed for drug-loaded microsphere formulations. These results suggest that drug was totally entrapped in the polymer.

Fig. 6: FTIR spectra.
a. Eudragit S100; b. drug; c. drug+Eudragit S100

Fig. 7: SEM images of various batches.
a. batch 3; b. batch 2; c. batch 16; d. batch 10; e. batch 8; f. batch 7; g. batch 6; h. batch 11

Fig. 8: DSC thermograms.
a. Clotrimazole drug; b. microspheres
Aggregates were not observed indicating uniformity of mass and a homogenous cream. pH of the cream was 7.05±0.03 which is nearly neutral, therefore non irritating to the skin. Clotrimazole drug content in the optimized cream was found to be 98.63±0.57%.
Spreadability of the given formulation is 30.56±0.18 g.cm/s which indicates good spreadability and viscosity was found to be 4485 cps at 20 rpm. Drug release was found to be very slow from polymer matrix in case of microsphere based cream as compared to conventional cream because of the retardation due to polymer used for microencapsulation of drug and complete drug release was achieved at the end of 24 h (fig. 9). Release of the drug from cream formulation using porcine ear skin i.e. ex vivo drug release was similar to that of in vitro release i.e. drug diffused very slowly from microsphere cream in comparison to conventional cream (fig. 10). Release was found to be 62.48±2.74% at the end of 10 h from microsphere cream whereas it was 88.63±3.46% from conventional cream. Drug permeated from conventional cream was high because drug in this formulation was in the free form for penetration whereas drug permeated from microsphere cream was low indicating increased accumulation of drug and formation of a depot or reservoir of the drug in the skin. As a result, decreased permeation into the diffusion fluid is attained. Flux and permeability coefficient was found to be 174.71 µg cm-2 h-1 and 0.05823 cm h-1, respectively. R2 value was found maximum in the case of zero-order graph, which clearly indicates that the formulations follow zeroorder release (Table 4) i.e. drug release is independent of its concentration present in the formulation. All the parameters related to stability of microsphere cream was found to be satisfactory at the end of three months which can be seen from the Table 5. The formulation did not show any changes in the physicochemical parameters over 3 months of storage at different temperature and humidity conditions. The viscosity of the cream was maintained within acceptable limits. Non-significant change in pH indicates the absence of drug degradation during storage. As observed, the long-term and accelerated data for any attribute did not show any significant change over time, hence apparent that the drug substance or product will remain well within the acceptance criteria during the proposed shelf life.

Fig. 9: Comparative in vitro drug release of microsphere cream and conventional cream.

Fig. 10: Comparative ex vivo drug release of microsphere cream and conventional cream.
| Formulation | Zero order (R2) | First order (R2) | Korsmeyerpeppas (R2) | Higuchi model (R2) | Flux (µg/cm2/h) |
Permeability coefficient (cm/s) | |
|---|---|---|---|---|---|---|---|
| In vitro | Conventional cream | 0.989 | 0.837 | 0.881 | 0.880 | 414 | 0.0138 |
| Microsphere cream | 0.980 | 0.970 | 0.825 | 0.926 | 318 | 0.0106 | |
| Ex vivo | Conventional cream | 0.993 | 0.853 | 0.852 | 0.891 | 394 | 0.0131 |
| Microsphere cream | 0.992 | 0.971 | 0.776 | 0.973 | 299 | 0.00996 |
Table 4: Release Kinetics
| Parameters | Initial | 1 month | 2 months | 3 months |
|---|---|---|---|---|
| Refrigeration condition (8±2°) | ||||
| Appearance | Smooth, opaque white | Smooth, opaque white | Smooth, opaque white | Smooth, opaque white |
| pH | 7.05 | 7.01 | 6.89 | 6.96 |
| Viscosity (cps) | 4485 | 4361 | 4299 | 4101 |
| % drug content | 98.63 | 98.76 | 98.23 | 98.05 |
| Room temperature (25±2°/60±5% RH) | ||||
| Appearance | Smooth, opaque white | Smooth, opaque white | Smooth, opaque white | Smooth, opaque white |
| pH | 7.05 | 7.11 | 7.03 | 6.99 |
| Viscosity (cps) | 4485 | 4328 | 4219 | 4113 |
| % drug content | 98.63 | 98.52 | 98.10 | 98.31 |
| Accelerated condition (40±2°/75±5% RH) | ||||
| Appearance | Smooth, opaque white | Smooth, opaque white | Smooth, opaque white | Smooth, opaque white |
| pH | 7.05 | 7.09 | 6.92 | 6.86 |
| Viscosity (cps) | 4485 | 4279 | 4298 | 4130 |
| % drug content | 98.63 | 98.06 | 97.84 | 97.27 |
Table 5: Stability Study
Skin irritation study conducted on Wistar albino rats when observed for sensitivity and reaction at the end of 24, 48 and 72 h showed absence of erythema or edema for both, the formulation and control as well. This indicates that prepared formulation does not cause any skin irritation with right choice of ingredients and so microsphere formula was suitable for human application with high safety and efficacy profiles. Results of in vitro antifungal activity are shown in fig 11. Initially, there was a sharp increase in the zone of inhibition in case of standard drug solution in DMSO and conventional cream as compared to microsphere cream after 8 h. This is because microspheres released drug slowly and less in amount. After 24 h the zone of inhibition was higher in the case of formulated systems clearly indicating that there was a continuous controlled release of drug from microsphere for a period of 24 h.

Fig. 11: Antifungal activity of formulation.
a. After 8 h; b. after 24 h
Microspheres were successfully prepared by optimizing various parameters and studying their effects on particle size as well as entrapment efficiency. It was then incorporated into a cream base to form a suitable topical drug delivery system. From the experiments, it was concluded that; as polymer amount increases, particle size and drug entrapment increase; as surfactant concentration increases, particle size and drug entrapment decreases; as stirring speed increases, particle size and entrapment decrease.
The microsphere loaded formulation of clotrimazole was developed to prolong the drug diffusion characteristics. Significant amount of the drug permeated to the receptor medium during both in vitro and ex vivo studies indicating efficient topical delivery of the drug through the membrane. Ex vivo diffusion studies showed that the drug followed zero order kinetics. The developed formulation was evaluated for physicochemical properties. FTIR spectroscopy of drug and polymer showed absence of drug polymer interactions. The formulation was found to be stable as per the stability studies conducted. No appreciable changes were observed in the physicochemical parameters at all storage conditions. Antifungal activity was evaluated and compared with conventional formulation indicating comparable antifungal activity. It is expected that, the topical side effects of the drug may be reduced and the efficiency of treatment may be increased as a result of prolonged release of drug from microparticles. Hence it can be concluded that the developed microsphere formulation is safe, efficacious and non-irritant and is promising in the treatment of fungal disease.
Acknowledgements
The authors thank Ciron Drugs Pvt. Ltd. for providing Clotrimazole drug as gift samples for this work. They also thank Dr. Mayur Yergiri for providing required facilities to carry out this research work at SVKM’s Dr. Bhanuben Nanavati College of Pharmacy, Mumbai. They are thankful to Tata Institute of Fundamental Research (TIFR), Mumbai for SEM analysis.
Financial Assistance
None.
Conflict of Interests
None declared.
References
- Kumar L, Verma S, Jamwal S, Vaidya S, Vaidya B. Polymeric microparticles-based formulation for the eradication of cutaneous candidiasis: Development and characterization. Pharm DevTechnol 2014;19:318-25.
- Khatry S, Sirish, Shastri N, Sadanandam M. Novel drug delivery systems for antifungal therapy. Int J Pharm PharmSci 2010;2:6-9.
- Badilli U, Sen T, Tarimci N. Microparticulate based topical delivery system of Clobetasol Propionate. AAPS PharmSciTech 2011;12:949-57.
- Basarkar G, Shirsath G, Patil S. Development of microspheres containing Diclofenacdiethylamine as sustained release topical formulation. Bull Pharm Res 2013;3:14-22.
- Labouta H, El-Khordagui L. Polymethacrylatemicroparticles gel for topical drug delivery. Pharm Res 2010;27:2106-18.
- Sudhamani T, Reddy N, Ravi Kumar V, Revathi R, Ganesan V. Preparation and evaluation of ethyl cellulose microspheres of ibuprofen for sustained drug delivery. Int J Pharm Res Dev 2010;2:119-25.
- Pavanveena C, Kavitha K, Anil S. Formulation and evaluation of trimetazidine hydrochloride loaded chitosan microspheres. Int J App Pharm 2010;2:11-4.
- Soni H, Singhai AK. Formulation and development of hydrogel based system for effective delivery of rutin. Int J App Pharm 2013;5:5-13.
- ICH, Q1A, Stability Testing of New Drug Substances and Products, in: Proceedings of theInternational Conference on Harmonisation, Geneva, October, 1993.
- Padmini R, Mallya R. Development and evaluation of a polyherbal topical formulation. JPR: Bio Med Rx: An Int J 2013;1:637-40.
- Helal DA, El-rhman DA, Abdel-halim SA, El-nabarawi MA.Formulation and evaluation of fluconazole topical gel. Int J Pharm PharmSci 2012;4:176-83.