- *Corresponding Author:
- Supriya Dixit
Biocontrol Laboratory, Department of Plant Pathology, Chandra Shekhar Azad University of Agriculture and Technology, Kanpur, Uttar Pradesh 208002, India
E-mail: supriya.dixit.28@gmail.com
| Date of Received | 17 July 2023 |
| Date of Revision | 17 May 2024 |
| Date of Acceptance | 20 August 2024 |
| Indian J Pharm Sci 2024;86(4):1499-1509 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
Tomato is one of the most cultivated and highly consumed crops globally, often hampered by a soil borne fungal pathogen Fusarium oxysporum f. sp. lycopersici causing vascular wilt. Pathogenic proteins like formyl peptide receptor 1, mitogen-activated protein kinase encoding, class V chitin synthase and tomatinase have been investigated in this study using molecular modeling, docking and dynamics-based techniques as potential molecular targets during pathogenesis or defence response. Thus, MODELLER 9.16 was used to predict three dimensional structures of the above proteins further refined and minimized. To get the new and dominant lead molecule, Trichoderma secondary metabolites were extracted from Trichoderma biomass isolated from healthy rhizospheric soil from an infected wilt field. The crude ethyl acetate extracts were purified and analyzed by gas chromatography-mass spectrometric analysis followed by in silico and in vitro analysis. Eventually, 85 compounds were selected in the first phase selection criteria for docking and it was found that molecule 1-allyl-4-[(3,4-dimethoxy-phenyl)-(1-thiophen-2-ylmethyl-1H-tetrazol-5-yl)-methyl]-piperazine and p-dihydroartemisinin oxymethyl benzoic acid were showed highest binding affinity towards Fusarium proteins as their binding energy fall in range of between -7.64 to -8.76 Kcal/mol. To increase the efficiency of molecules the five derivatives were designed and based upon the docking results, three potential derivates undergone for molecular dynamic studies over 100 ns against pathogenic proteins. Thus, aiding future development of the designed potent derivatives can be explored for experimental validation against the Fusarium pathogenic proteins to complement the current management techniques. This improvised new strain of Trichoderma species would assist in controlling disease effectively, especially against soil-borne pathogens.
Keywords
Tomato plant, fungal pathogens, protein sequencing, Fusarium species, Trichoderma, molecular modeling, secondary metabolite, molecular docking, biochemical analysis
Fungal plant pathogens cause various diseases in plants which may lead to reduced crop quality and yield and commercial crop losses are mostly caused by systemic foliar infections. In this way, it follows the destruction of forests and agriculture[1]. As a result, they are essential to our way of life in terms of environmental sustainability, economic prosperity and food safety. Fusarium sp. represent the most intricate and diverse group of both pathogenic and non-pathogenic fungi[2]. Fusarium toxins are thought to be the most prevalent natural contaminants of food grains that cause several diseases in humans, as they are well capable of producing several toxic enzymes that cause major devastating diseases like vascular wilt, blight, rotting of fruits and roots, leaf spots, crackers, etc., to various economically important food crops[3]. Fusarium oxysporum f. sp. lycopersici (Fol) over 100 different forma speciales (including pathogenic and non-pathogenic species), causes vascular wilt of the tomato which is the edible fruit cultivated globally as an annual crop[4].
In India, tomato is the second largest vegetable crop with a rich source of antioxidants and micronutrients that promote good health and also help to reduce the risk of cardiovascular disease and cancer[5]. Despite many varieties of tomatoes being developed through genetic improvement, the Fusarium wilt tomato is one of the most widespread and disastrous diseases that causes great yield loss, specifically in warm climates and temperate regions[6].
Several chemical-based fungicides, carbendazim, metalaxyl and mancozeb have been used to control Fusarium wilt[7]. Biological control is the best alternative for the management of wilt disease because chemicals have a huge negative impact on the environment and also cause pollution[8]. Nowadays, many biocontrols have been used and Trichoderma species are one of them, known for their efficacy against various broad spectrums of plant pathogens[9,10] by their mycoparasitism mechanism. It involves various steps to recognize the host, attack the pathogen and then eliminating it. During this process, they produce a higher number of secondary metabolites which play a vital role. They are a group of chemically distinct natural and heterologous lower molecular weight substances. It also includes various biological activities like antibiotic, antifungal and anticancer, which are not only capable of inhibiting the growth of other micro/macro-organisms but also regulate growth and yield of plants without causing any further harmful effect to humans. Dixit et al.[11], identified Fusarium virulent genes causing virulence in tomato plants and humans but their three Dimensional (3D) structures are not available yet. Hence, this work aims to focus on pathogenic proteins that will serve as molecular targets of Fol and it is possible to find significant lead Trichoderma secondary metabolite compounds that can neutralize the aforementioned proteins during pathogenesis by using molecular docking and dynamics techniques. This new methodology is an attempt through the application of in silico techniques to design potential lead derivatives that can protect tomato plants from Fol. Further Trichoderma bioformulations were tested via in vitro techniques to improve and promote the growth and yield of tomatoes without degrading their quality (e.g., taste, smell) and used as an alternative of chemical-based fertilizer.
Materials and Methods
Sequence retrieval, structure prediction and refinement:
Identification and protein sequence retrieval of Fusarium wilt-causing genes encoding proteins were selected based on the previous study done by Dixit et al.[11]. The protein sequence of related Pathogenesis-Related 1 (PR-1) (ACV31371.1), mitogen-activated protein kinase encoding (fmk1) (AAG01162.1), class V chitin synthase (chsV) (AAO49384.1) and tomatinase (tom1) (CAA10112.1) were obtained from National Center for Biotechnology Information (NCBI). Their 3D structures were not available hence; the prediction of the 3D model was planned accordingly. BLASTP searches closely related homologs sequences were chosen and modeled via comparative modeling through MODELLER 9.16[12]. Models with high DOPE and low molecular PDF (molpdf) were selected and the stereochemical properties of each model were checked by the PROCHECK server[13]. Functional Gene-Module Detection (FG-MD) and ModRefiner servers both are available at https://zhanglab.ccmb.med.umich.edu/ and were selected for energy minimization and refinement of predicted 3D structures. FG-MD is a molecular dynamics program for 3D structure refinement according to close template structure[14] whereas ModRefiner server is a high-resolution protein structure refinement algorithm that improves physical quality structures that are close to their native state[15].
Extraction, identification and evaluation of secondary metabolites:
Six potential Trichoderma isolates Trichoderma asperellum (T. asperellum), Trichoderma longibrachiatum, and Trichoderma koningii (T. koningii) were taken from the Biocontrol Lab, Chandra Shekhar Azad University of Agriculture and Technology, Kanpur (Sourced from the state of Uttar Pradesh, India) for the identification of natural secondary metabolites which will be further used as ligands to inhibit Fusarium pathogenic proteins.
Trichoderma isolates were grown in potato dextrose broth and incubated at 28° for 25 d. Afterward, the content of flasks was filtered through muslin clothes and the obtained liquid phase was used for the extraction of volatile/non-volatile metabolites. The solvent extraction method was used for metabolite extraction, in which metabolites were mixed into ethyl acetate at a ratio of 1:1 (v/v). The upper phase of the solvent which contains metabolites was collected through the separating funnel into conical flasks. Ethyl acetate was evaporated from the collected upper phase and the remaining residues were dissolved in acetone which was used for Gas Chromatography-Mass Spectrometric (GC-MS) analysis[16]. 3D structures of Trichoderma secondary metabolites retrieved from the PubChem database have proceeded for selection of the set of ligands based on the calculation of bioactivity and drug-likeness properties using Molinspiration (www.molinspiration.com) and their physiochemical properties check via Swiss- Absorption, Distribution, Metabolism, and Excretion (SwissADME) server[17].
Molecular docking and dynamics:
Identification of active sites of the targets is the initial step before heading for docking and was obtained by using the Active Site Prediction server (http://www.scfbio-iitd.res.in/dock/ActiveSite. jsp). The cavity with the highest volume was selected. AutoDock software[18] is used for docking analysis; it uses the Lamarckian Genetic Algorithm (LGA) to pick out one of the best conformations of ligands within the active site of targets. As the software is compatible with the PDBQT file format, so all proteins and ligands files were prepared accordingly and the grid box size of receptors was determined by AutoDock Tools (ADT). Further, the best-docked ligands molecules were selected and their modified derivatives were drawn by MarvinSketch (http://www.chemaxon. com/products/marvin/marvinsketch/) software[19].
Based on the docking score, complexes were selected and underwent Molecular Dynamics Simulation (MDS) for 100 ns by employing AMBER18. MDS was performed to analyze the stability of complexes concerning the physical change of macromolecules. It also calculates the strength, dynamic conformational change and intermolecular properties by evaluating Root Mean Square Deviation (RMSD), intermolecular hydrogen bond interactions, Root Mean Square Fluctuations (RMSF), binding free energy and Radius of Gyration (RoG) for the elucidation of conformational, structural and compactness of the protein-ligand[20]. To start with MDS, the first software’s leap module created the molecular receptor topology, followed by neutralization and submerging of the query complex into the solvent with a rectangular box (TIP3P). The distance between the solvent boxes to the macromolecules was fixed at 10 Å. The minimization procedure of the solvent system is accomplished by fixing the protein with the restraint of the heavy atoms by removing bad contacts, initially, 2500 steps of the steepest descent method were performed which were followed by 2000 steps of conjugation gradient. Next step, the system starts to heat up from 0°K-300°K temperature at 1 atm pressure for 50 ps followed by density equilibrium of 50 ps and 1 ns of constant equilibrium, and the temperature was maintained by using the Berendsen algorithm. Then MDS was run for 100 ns to evaluate the stability of docked protein-ligand complexes at 500 ps system equilibration on canonical constanttemperature, constant-pressure (NTP) ensemble. The covalently bound hydrogen atoms were constrained by the SHAKE algorithm with 2 fs time and temperature maintained according to Langevin dynamics. The final recording was made for every 5 ps by employing the particle-mesh Ewald summation method. After 100 ns of simulation, the results were analyzed by the CPPTRAJ module of the AMBER18 in terms of RMSD, RMSF and RoG. Finally, Molecular Mechanics with Generalized Born and Surface Area (MM-GBSA) binding free energy was calculated.
Biochemical analysis:
To test the effect of volatile and non-volatile compounds of Trichoderma isolates, the tomato seeds were treated with crude and talc base formulations of Trichoderma isolates against Fol. Tomato plants were grown under in vitro conditions for 21 d, after germination, leaf samples were taken from all treatments and subjected to various biochemical analyses. Total chlorophyll content in tomato plants was determined according to Arnon et al.[21]. Similarly, peroxidase activity with dehydrogenation of guaiacol and Polyphenol Oxidase (PPO) activity with 4-methylcatechol as substrates were assayed[22,23]. Total phenolic content was estimated by using Folin-Ciocalteu reagent[24].
Statistical analysis:
The treatments were compared using Critical Differences (CD) at 5 % significance level.
Results and Discussion
Since the 3D structure of Fol pathogenic proteins was not solved experimentally, the 3D structure prediction and modeling of pathogenic proteins were determined through homology modeling through Basic Local Alignment Search Tool (BLAST) search followed by intensive optimization and validation. For accurate modelling of target sequences, it is necessary to choose a reliable template structure that is more than 50 % of sequence similarity and also has a resolution of <2 ?. Further models were developed by MODELER 9.19 and their stereo-chemical assessment was followed by PROCHECK webserver. The stereochemistry analysis of the targets revealed >80 % of residues are in favored regions of Ramachandran plot. To get more reliable and stable structures, refinement and minimization of the predicted 3D models were assessed by two web servers, FG-MD and ModRefiner simultaneously obtained final structures were re-evaluated through PROCHECK and were compared to the previous one (Table 1 and fig. 1).
| S. no | Protein name | Before Refinement | After Refinement | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Favored regions (%) | Allowed regions (%) | Generously allowed regions (%) | Disallowed regions (%) | Favored regions (%) | Allowed regions (%) | Generously allowed regions (%) | Disallowed regions (%) | ||
| 1 | pr1 | 82.9 | 11.6 | 3.7 | 1.9 | 87 | 10.2 | 1.4 | 1.4 |
| 2 | fmk1 | 89.4 | 8.8 | 1.2 | 0.6 | 90.6 | 7 | 1.9 | 0.6 |
| 3 | tom1 | 86.5 | 11.8 | 1.0 | 0.7 | 88.2 | 10.4 | 1 | 0.3 |
| 4 | CVS | 90.2 | 7.4 | 1.5 | 0.9 | 92.8 | 6.6 | 0.4 | 0.2 |
Table 1: Ramachandran Plot Results (Core Region) of 3D Structure Modelled by Refinement Program

Fig. 1: Structures, (A): PR-1; (B): fmk1; (C): chsV and (D): tom1 proteins of Fol
The important step of the present research work is to identify group of natural lead compounds to inhibit the growth of targets and increase the production of crops. So, the results of partly purified crude extract GC-MS analysis yield more than 300 compounds with molecular mass ranging from 162.14-937.82 g/mol. Compounds with a high percentage of occurrences were selected for the prediction of drug-likeness properties, to eliminate compounds having poor chemical and physical properties also insoluble and nonpermeable. From the observation, it was found that only 85 compounds were found to obey Lipinski’s rule[25].
The active sites of the Fusarium pathogenic proteins were obtained by using the active site prediction server (http://www.scfbio-iitd.res.in/ dock/ActiveSite_new.jsp), it identifies different cavities of a protein along with their respective XYZ coordinates and highest cavity volume were selected and compounds were docked using AutoDock software. Grid box was generated near the binding site of a protein and a Lamarckian genetic algorithm was selected to generate the best conformers. The docked ligands were selected based on the docking score with the best conformation. Out of the selected 85 compounds, only two compounds were bound potentially. The first compound is 1-allyl-4-[(3,4-dimethoxyphenyl)-( 1-thiophen-2-ylmethyl-1H-tetrazol-5- yl)-methyl]-piperazine, which interacted with PR-1, tom1 and chorionic villus samples proteins. The second compound is p-dihydroartemisinin oxymethyl benzoic acid, which interacted with fmk1 protein with the least docking score (Table 2, Table 3 and fig. 2).
| S. no | Secondary metabolites | pr1 | fmk1 | tom1 | chsV |
|---|---|---|---|---|---|
| 1 | 1-allyl-4-[(3,4-dimethoxy-phenyl)-(1-thiophen-2-ylmethyl-1H-tetrazol-5-yl)-methyl]-piperazine | -7.64 Kcal/mol | -6.50 Kcal/mol | -8.54 Kcal/mol | -8.09 Kcal/mol |
| 2 | p-dihydroartemisininoxymethylbenzoic acid | -6.88 Kcal/mol | -8.76 Kcal/mol | -7.20 Kcal/mol | -5.57 Kcal/mol |
Table 2: Molecular Docking of Screened Compound against Targeted Proteins
| S. no | Protein-ligand complex | Docking energy | Interacting residue | Docked structures |
|---|---|---|---|---|
| 1 | pr1 and derivative 05 | -8.37 | GLN110 | 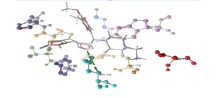 |
| 2 | chsV and derivative 03 | -8.27 | ASP182 | 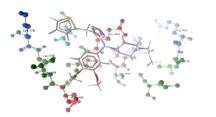 |
| 3 | tom1 and derivative 05 | -10.03 | GLU259, TRP292 | 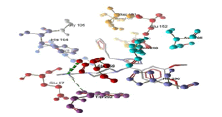 |
| 4 | fmk1 and pDerivative 05 | -9.25 | TYR185 | 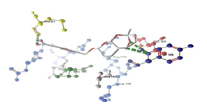 |
Table 3: Docking analysis

Fig. 2: Structure of top derivatives of 1-allyl-4-[(3, 4-dimethoxy-phenyl)-(1-thiophen-2-ylmethyl-1H-tetrazol-5-yl)-methyl]-piperazine and p-dihydroartemisininoxymethylbenzoic acid
The physicochemical properties of topdocked designed derivatives were predicted by SwissADME server to evaluate the drug-likeness (Table 4). From the docking studies, it was confirmed that out of 85, only two compounds are best fitted into the modeled structures and the qualified leads were taken further to check their efficacy via molecular simulation (fig. 3).
| S. no | Physicochemical properties | Derivative 05 | Derivative 03 | pDerivative 05 |
|---|---|---|---|---|
| 1 | Formula | C20H27N7O2S | C22H30N6O2S | C23H31NO7 |
| 2 | Molecular weight | 429.54 g/mol | 442.58 g/mol | 433.49 g/mol |
| 3 | Num. heavy atoms | 30 | 31 | 31 |
| 4 | Num. arom. heavy atoms | 16 | 16 | 6 |
| 5 | Fraction Csp3 | 0.45 | 0.5 | 0.7 |
| 6 | Num. rotatable bonds | 8 | 9 | 4 |
| 7 | Num. H-bond acceptors | 8 | 7 | 7 |
| 8 | Num. H-bond donors | 1 | 0 | 2 |
| 9 | Molar refractivity | 122.13 | 129.04 | 111.92 |
| 10 | TPSA | 122.80 Å2 | 96.78 Å | 109.47 Å2 |
| 11 | Log Po/w | 1.56 | 2.95 | 2.92 |
Table 4: Physiochemical Properties of Top Docked Designed Derivatives

Fig. 3: MDS results at 100 ns, (A): RMSD; (B): RMSF; (C): Inter-molecular hydrogen bonding (H bonding) and (D): RoG
The potential isolates i.e., T. koningii and T. asperellum crude and talc formulations were prepared and checked for their ability to stimulate Phenol Oxidase (PO) and PPO activities in the tomato. To find the PO, PPO, phenols activity and chlorophyll content were evaluated against test fungi Fol in vitro. The data were arranged statistically and the results are presented below in Table 5.
| S. no | Treatment | Peroxidase activity | Polyphenol oxidase activity | Phenol content |
|---|---|---|---|---|
| 1 | T. koningii (Talc) | 303.95 | 0.0872 | 94.93 |
| 2 | T. koningii (Crude) | 274.28 | 0.0366 | 92.17 |
| 3 | T. asperellum (Crude) | 279.35 | 0.0607 | 72.14 |
| 4 | T. asperellum (Talc) | 232.09 | 0.0604 | 65.74 |
| 5 | Control | 121.00 | 0.0124 | 22.01 |
| 6 | Negative control | 154.51 | 0.0212 | 34.26 |
| 7 | SE (d) | 1.576 | 0.0017 | 0.451 |
| 8 | CD at 5 % | 3.435 | 0.0037 | 0.984 |
Table 5: Efficacy of Trichoderma Treated Seeds in PO, PPO and Phenol Activity against Fusarium Oxysporum F. Sp. Lycopersici
The results of biochemical activity were significantly higher in seed treated with talc formulation of T. koningii as compared to control followed by the crude formulation of T. asperellum against Fol, the highest PO, PPO and phenol content were observed in seed treated with T. koningii (Talc).
The total Chl content was recorded in the above nursery experiment in the 3rd w after germination and the results are summarized in Table 6. The below result indicated that the highest Chl content was also observed in seeds treated with talc formulation of T. koningii followed by the crude formulation of T. asperellum.
| S. no | Treatment | Chlorophyll content | ||
|---|---|---|---|---|
| Fol | ||||
| Chl-A | Chl-B | Total | ||
| 1 | T. koningii (Talc) | 1.93 | 0.42 | 2.59 |
| 2 | T. koningii (Crude) | 1.35 | 0.65 | 2 |
| 3 | T. asperellum (Crude) | 1.14 | 0.65 | 1.56 |
| 4 | T. asperellum ( Talc) | 1.23 | 0.24 | 1.47 |
| 5 | Control | 0.58 | 0.25 | 0.83 |
| 6 | Negative control | 1.04 | 0.36 | 1.4 |
| 7 | SE(d) | 0.0642 | 0.022 | 0.075 |
| 8 | CD at 5 % | 0.1399 | 0.049 | 0.165 |
Table 6: Effect of Seed Treatment with Trichoderma Isolates on Total Chlorophyll Content in Tomato Seedlings
Fusarium wilt is a very serious disease caused by soil-inhabiting fungus Fol f. sp. lycopersici, is one of the most devastating diseases of tomato. This disease is characterized by yellowed leaves and wilted plants with minimal crop yield. Approximate 30 %-40 % yield loss due to wilt disease and increased up to 80 % under favorable weather conditions. Whole-genome sequencing and proteomics studies identified many diseasecausing pathogenic proteins and enzymes including pr1, fmk1, chV and tom1 enzymes, all of which play a key role in pathogenesis of Fol in both plants and humans.
However, the 3D structures of above said targets were not experimentally driven so, it would become comparatively easy to predict the 3D structure by homology modeling, high-resolution models of Fol were predicted and their stereochemical quality was validated through PROCHECK. Similar work was performed in 2012 by Rai et al.[26]; they predicted and analyzed the 3D structure of the Bovine rhinitis B virus RNA dependent RNA polymerase via comparative modelling through SWISS-MODEL and Geno3D and further verified its accuracy through PROCHECK, further they make a comparison between predicted and crystal structures. It was observed that the SWISSMODEL gives the best and most accurate model. Another study was conducted by Prajapati et al.[27], Clostridium botulinum is a pathogenic bacterium but the crystal structures of pathogenic protein were not available in Protein Data Bank (PDB) hence they predicted the homology model via SWISS-MODEL and MODELLER and validated through PROCHECK the predicted models are the foundation for future functional analysis.
Traditionally homology-built models rely on template proteins that possess precise stereochemistry as this method simply copies the structure information from the template protein by aligning the similar regions of the query protein. Therefore, the predicted structures were more similar to the template protein rather than the native structure. Although the predicted structure in this manner often leads to a significant error where proper alignment cannot takes place. These regions majorly include loops, non-conserved regions of side chains and insertion of any length. To solve the above questions, refinement and minimization of homology-based predicted 3D structures are the crucial steps as they improve structural quality in terms of favorable stereochemistry, stability and avoidance of clashes that lead to predicting approximate experimental accurate structures close to their native form.
Enormous previous studies revealed that several ecological pest controls and biofertilizers are used but among them, Trichoderma is one of the most potential fungal biocontrol agents that is used frequently as a biopesticide worldwide chosen for this study to find a new set of natural compounds against Fusarium species because fungal secondary metabolites are low weight molecules which can easily penetrate fungal/plants cell walls, stimulates biocontrol activity by activating specific mycoparasitism related genes. In 2020, Moutassem et al.[28], investigated the effects of five Trichoderma sp. Samarium (SM) extracted by solvent extraction methods against Fusarium oxysporum f. sp. ciceris, in vitro test results showed significant inhibitory effects upto 73.8 % and 27.8 %, also in vivo test showed that secondary metabolites actively reduce disease severity. Hence SMs not only exhibit a broad class of antifungal, antibiotics, antiprotozoal and anticancer activity but also regulate systemic defense resistance and promote plant growth[29]. Vinale et al., state that more than 40 various secondary metabolites secreted that have phytopathogenic action, which includes volatile and non-volatile substances such as 6-n-pentyl-6H-pyran-2-one (6PP), harziandione, viridin, harzianopyridone, gliotoxin and peptaibols[30].
There are various agriculturally important natural compounds are found abundantly and along with in silico techniques it becomes easier to discover and design new agrochemicals by implanting computer-aided drug designing which avails raw experimental data and turns it into attainable data. Nowadays various agriculture and pharmaceutical industries efficiently use computational approaches to screen optimize and evaluate the activity of the natural compounds by minimizing the cost and time at the same time[31]. A molecular docking study reveals that 1-allyl-4-[(3,4-dimethoxyphenyl)-( 1-thiophen-2-ylmethyl-1H-tetrazol-5- yl)-methyl]-piperazine and p-dihydroartemisinin oxymethyl benzoic acid are two most potential SM against Fusarium pathogenic proteins. So, based on our first observations, five derivatives were designed by adding OH, OCH, CH3, COOH and NH2 functional groups to both compounds shown in (fig. 2), no replacements were done in structures because the other part is involved in protein-ligands interaction hence only addition of functional groups done to derive derivatives with improvised binding affinity towards pathogenic proteins and re-docked and final docking results showed increased affinity of compounds having CH3 and NH2 group shown in (Table 3). A similar work was done by Pathak et al.[32], where they docked four phytoalexins against molecular targets of Alternaria brassicicola and found spirobrassinin is best docked with all targets and to improve their compounds binding affinity, they designed five derivatives by replacing RCH3 group to OH and CH3 and docked again, it was found that derivative spirobrassinin 02 and 05 (OH and ROCH3, respectively) have high binding affinity.
The physicochemical properties of top-docked designed derivatives were predicted by SwissADME server to evaluate the drug-likeness (Table 4). Further bioactivity of identified compounds was searched through the Pubchem database, it was found that compound 1-allyl-4-[(3,4-dimethoxyphenyl)-( 1-thiophen-2-ylmethyl-1H-tetrazol- 5-yl)-methyl]-piperazine which is identified from T. asperellum was tested as an inhibitor against TEAD-YAP interaction, microphthalmiaassociated transcription factor, one of the factors that cause skin cancer[33] and activator of GPR151, E3 ligase (FBW7) found inactive in all tests while p-dihydroartemisinin oxymethyl benzoic acid from T. koningii is artemisinin which is used as antimalarial drug have but studies found that it has potent anti-inflammatory and immunoregulatory properties, as well as the ability to regulate oxidative stress besides having all these properties, Xia et al., patent the compound and its derivatives were for the treatment of kidney diseases[34].
In the dynamics study the parameters explored for analyses such as RMSD, RMSF, inter-molecular H bonding, RoG and binding free energy studied and dynamics were performed for all complexes, results of MDS analysis showed higher stability found throughout the dynamics run was in between pr1 with 1-allyl-4-[(3,4-dimethoxy-phenyl)-(1- thiophen-2-ylmethyl-1H-tetrazol-5-yl)-methyl]- piperazine secondary metabolite while other complexes result found to be highly fluctuating.
Through RMSD analysis, the stability and behavior of ligand-ligand complexes were calculated in a defined time while RMSF gives information on the conformational flexibility of the protein structure by describing its flexible and rigid regions[35]. The RMSD and RMSF values are described in fig. 3. Here in this study, MDS has performed over 100 ns. RMSD graph depicted that the ligand was initially fluctuating but will successively reach stabilization at 70 ns and will remain the same till 100 ns. Thus the ligand was stabilized during most time of the dynamics run whereas RMSF graph showed fluctuations in the loop region of the complex, representing the generous nature of the binding site of proteins represent that the ligand can be generously bound to protein. The binding affinity of the ligand is determined by the number of hydrogen bonds formed between ligand-protein interactions. The stronger the binding affinity, the more hydrogen bonds there are in the complex. The formation and disruption of hydrogen bonds are therefore considered by MDS analysis. The present study observes three hydrogen bonds at the end of 100 ns simulation.
RoG is being used to evaluate the compactness and folding of protein-ligand complexes. It is a key method for proving conformational structural changes in protein after ligand interaction. The higher value of RoG illustrates loose and weak interaction formation in protein after interaction with ligand. Considering the following points, RoG graph depicts that there were not many variations observed throughout the 100 ns and the recoded average high value of around 20 Å which shows that the ligand is tightly bound to the active site of the protein. Total binding free energy (ΔG) was also calculated in terms of MM/GBSA and -35.2900+5.3617 Kcal/mol which falls into the favorable range.
The molecular modeling and docking analysis showed that molecule 1-allyl-4-[(3,4-dimethoxyphenyl)-( 1-thiophen-2-ylmethyl-1H-tetrazol-5- yl)-methyl]-piperazine and p-dihydroartemisinin oxymethyl benzoic acid and its derivatives are potential compounds, molecular dynamics study reveals that the above-said compounds are in stable form of Fol pathogenic proteins. To our knowledge, this is the first research to confirm the presence of both compounds used as anthelmintic drugs in Trichoderma species while in vitro biochemical studies showed that talc formulations of T. koningii have significantly higher PO, PPO, phenols activity and chlorophyll content against Fol. The study also suggests that T. koningii talc-based formulations can be used as a natural substitute in place of chemical-based treatment that causes damage to tomato plants and the environment and increases growth and yield. The present study reports that p-dihydroartemisinin oxymethyl benzoic acid is identified in T. koningii as a compound is used to treat malaria and renal disease. Further, experimental validation is required to confirm their potency and efficacy for further synthesis and development for the usage in broad-spectrum.
Acknowledgments:
The authors are thankful to Mr. Dheeraj Kumar Chaurasia, Supercomputing Facility for Bioinformatics and Computational Biology, Indian Institute of Technology, Delhi for support during MDS studies. The authors are thankful to the Indian Institute of Science Education and Research (IISER), Bhopal for GC-MS analysis.
Authors’ contributions:
Study conception and design was done by Supriya Dixit, Mukesh Srivastava, Pramod Katara; data collection was done by Supriya Dixit; analysis and interpretation of results was done by Supriya Dixit, Mukesh Srivastava, Pramod Katara; resources was done by Mukesh Srivastava, Pramod Katara and manuscript was prepared by Supriya Dixit, Mukesh Srivastava, Pramod Katara. All authors reviewed the results and approved the final version of the manuscript.
Conflict of interests:
The authors declared no conflict of interests.
References
- Jain A, Sarsaiya S, Wu Q, Lu Y, Shi J. A review of plant leaf fungal diseases and its environment speciation. Bioengineered 2019;10(1):409-24.
[Crossref] [Google Scholar] [PubMed]
- Gordon TR. Fusarium oxysporum and the Fusarium wilt syndrome. Annu Rev Phytopathol 2017;55(1):23-39.
[Crossref] [Google Scholar] [PubMed]
- Srinivas C, Devi DN, Murthy KN, Mohan CD, Lakshmeesha TR, Singh B, et al. Fusarium oxysporum f. sp. Lycopersici causal agent of vascular wilt disease of tomato: Biology to diversity–A review. Saudi J Biol Sci 2019;26(7):1315-24.
[Crossref] [Google Scholar] [PubMed]
- Akaeze OO, Aduramigba-Modupe AO. Fusarium wilt disease of tomato: Screening for resistance and in vitro evaluation of botanicals for control; the Nigeria case. J Microbiol Biotechnol Food Sci 2017;7(1):32.
- Kumar KS, Paswan S, Srivastava S. Tomato-a natural medicine and its health benefits. J Pharmacog Phytochem 2012;1(1):33-43.
- Agrios GN. Plant pathology. Elsevier 2005.
- Sitara U, Hasan NU. Studies on the efficacy of chemical and non chemical treatments to control mycoflora associated with chilli seed. Pak J Bot 2011;43(1):95-110.
- Mohiddin FA, Khan MR, Khan SM, Bhat BH. Why Trichoderma is considered Super Hero (Super fungus) against the evil parasites? Plant Pathol J 2010;9:92-102.
- Harman GE, Howell CR, Viterbo A, Chet I, Lorito M. Trichoderma species—opportunistic, avirulent plant symbionts. Nat Rev Microbiol 2004;2(1):43-56.
[Crossref] [Google Scholar] [PubMed]
- Chet I, Harman GE, Baker R. Trichoderma hamatum: Its hyphal interactions with Rhizoctonia solani and Pythium spp. Microb Ecol 1981;7:29-38.
- Dixit S, Srivastava M, Katara P. Computational assessment of polymorphism with linkage disequilibrium and hotspots of recombination in pathogenic genes of Fusarium oxysporum f. sp. Lycopersici. Legume Res Int J 2019;42(6):856-61.
- Šali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993;234(3):779-815.
[Crossref] [Google Scholar] [PubMed]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallograp 1993;26(2):283-91.
- Zhang J, Liang Y, Zhang Y. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure 2011;19(12):1784-95.
[Crossref] [Google Scholar] [PubMed]
- Xu D, Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J 2011;101(10):2525-34.
[Crossref] [Google Scholar] [PubMed]
- Sonika Pandey SP, Mukesh Srivastava MS, Mohammad Shahid MS, Anuradha Singh AS, Shubha Trivedi ST, Vipul Kumar VK, et al. Isolation and identification of volatile metabolites from the biocontrol agent Trichoderma harzianum through GC-MS analysis.
- Daina A, Michielin O, Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecule. Sci Rep 2017;7(1):42717.
[Crossref] [Google Scholar] [PubMed]
- MarvinSketch. ChemAxon; 2023.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 2009;30(16):2785-91.
[Crossref] [Google Scholar] [PubMed]
- Yadav R, Imran M, Dhamija P, Chaurasia DK, Handu S. Virtual screening, ADMET prediction and dynamics simulation of potential compounds targeting the main protease of SARS-CoV-2. J Biomol Struct Dyn 2021;39(17):6617-32.
[Crossref] [Google Scholar] [PubMed]
- Arnon DI. Copper enzymes in isolated chloroplasts. Polyphenoloxidase in Beta vulgaris. Plant Physiol 1949;24(1):1.
[Crossref] [Google Scholar] [PubMed]
- Malik CP, Singh MB. Mallik CP, Singh MB. Plant enzymology and histoenzymology. Text Manual 1980;434.
- Zauberman G, Ronen R, Akerman M, Weksler A, Rot I, Fuchs Y. Post-harvest retention of the red colour of litchi fruit pericarp. Sci Horticulturae 1991;47(1-2):89-97.
- Ainsworth EA, Gillespie KM. Estimation of total phenolic content and other oxidation substrates in plant tissues using Folin–Ciocalteu reagent. Nat Protoc 2007;2(4):875-7.
[Crossref] [Google Scholar] [PubMed]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 1997;23(1-3):3-25.
[Crossref] [Google Scholar] [PubMed]
- Rai DK, Rieder E. Homology modeling and analysis of structure predictions of the bovine rhinitis B virus RNA dependent RNA polymerase (RdRp). Int J Mol Sci 2012;13(7):8998-9013.
[Crossref] [Google Scholar] [PubMed]
- Prajapati C, Bhagat C. In-silico analysis and homology modeling of target proteins for Clostridium botulinum. Int J Pharm Sci Res 2012;3(7):2050.
- Moutassem D, Belabid L, Bellik Y. Efficiency of secondary metabolites produced by Trichoderma spp. in the biological control of Fusarium wilt in chickpea. J Crop Prot 2020;9(2):217-31.
- Vinale F, Sivasithamparam K, Ghisalberti EL, Woo SL, Nigro M, Marra R, et al. Trichoderma secondary metabolites active on plants and fungal pathogens. Open Mycol J 2014;8(1).
- Vinale F, Sivasithamparam K, Ghisalberti EL, Ruocco M, Woo S, Lorito M. Trichoderma secondary metabolites that affect plant metabolism. Nat Product Commun 2012;7(11):1934578X1200701133.
[Google Scholar] [PubMed]
- Lamberth C, Jeanmart S, Luksch T, Plant A. Current challenges and trends in the discovery of agrochemicals. Science 2013;341(6147):742-6.
[Crossref] [Google Scholar] [PubMed]
- Pathak RK, Taj G, Pandey D, Kasana VK, Baunthiyal M, Kumar A. Molecular modeling and docking studies of phytoalexin (s) with pathogenic protein (s) as molecular targets for designing the derivatives with anti-fungal action on Alternaria spp. of Brassica. Plant Omics 2016;9(3):172-83.
- Wang J, Fang P, Chase P, Tshori S, Razin E, Spicer TP, et al. Development of an HTS-compatible assay for discovery of melanoma-related microphthalmia transcription factor disruptors using alphaScreen technology. SLAS Discov 2017;22(1):58-66.
[Crossref] [Google Scholar] [PubMed]
- Xia M, Liu D, Liu Y, Liu H. The therapeutic effect of artemisinin and its derivatives in kidney disease. Front Pharmacol 2020;11:380.
[Crossref] [Google Scholar] [PubMed]
- Kesharwani A, Chaurasia DK, Katara P. Repurposing of FDA approved drugs and their validation against potential drug targets for Salmonella enterica through molecular dynamics simulation. J Biomol Struct Dyn 2022;40(14):6255-71.
[Crossref] [Google Scholar] [PubMed]