- *Corresponding Author:
- A. Gorajana
School of Pharmacy and Health Sciences, International Medical University, Bukit Jalil, 57000, Kuala Lumpur, Malaysia
E‑mail: glknarayana@yahoo.com
| Date of Submission | 23 March 2012 |
| Date of Revision | 05 March 2013 |
| Date of Acceptance | 17 March 2013 |
| Indian J Pharm Sci 2013;75(3):291-301 |
Abstract
The aim of this study was to enhance the dissolution rate of efavirenz using solid dispersion systems (binary and ternary). A comparison between solvent and fusion method was also investigated. Solid dispersions of efavirenz were prepared using polyethylene glycol 8000, polyvinylpyrrolidone K30 alone and combination of both. Tween 80 was incorporated to obtain a ternary solid dispersion system. Dissolution tests were conducted and evaluated on the basis of cumulative percentage drug release and dissolution efficiency. Physicochemical characterizations of the solid dispersions were carried out using differential scanning calorimetric, powder X-ray diffraction, Fourier transform infrared spectroscopy, and scanning electron microscopy. Dissolution was remarkably improved in both systems compared to pure efavirenz ( P<0.05). An optimum ratio was identified at a drug:polymer of 1:10. Incorporation of Tween 80 to 1:10 formulations formed using solvent method showed further improvement in the dissolution rate. Physicochemical characterization results suggested that efavirenz existed in the amorphous form in all the solid dispersion systems providing evidence of improvement in dissolution. No statistically significant difference ( P>0.05) in dissolution was observed between the two methods. Binary and ternary solid dispersion systems both have showed a significant improvement in the dissolution rate of efavirenz. Formulations with only polyvinylpyrrolidone K30 showed best dissolution profile and 1:10 was identified as an optimum drug-polymer weight ratio.
Keywords
Solid dispersion, efavirenz, dissolution enhancement, polyethylene glycol, PVP K30, Tween 80
The oral route of drug administration is the most common and preferred method of delivery. However, several orally administered drugs have a reduced bioavailability due to poor water solubility. In biopharmaceutical classification system drugs with low aqueous solubility, slow dissolution rate, high dose, and high membrane permeability are categorized as Class II drug [1]. To overcome low bioavailability, many of the modern oral drug delivery systems emphasize on formulation strategies such as alteration of solvent composition, carrier systems as well as chemical and physical modifications [2]. Solid dispersion of drug in a water soluble polymer has been shown to be one of the most promising strategy to improve solubility [3]. Polyethylene glycols (PEG) are polymers of ethylene oxide, with a molecular weight (MW) usually falling in the range 200-300 000. For the manufacture of solid dispersions and solutions, PEGs with molecular weights of in the range of 1500 to 20 000 are usually employed.
As the MW increases, so does the viscosity. They are most commonly used because of their good solubility in water and in many organic solvents, low melting points (under 65°), ability to solubilize some compounds, and improvement of compound wettability. The relatively low melting point is advantageous for the manufacture of solid dispersions by the melting method. PEG 8000 is a hydrophilic polymer that has been used in the preparation of solid dispersion systems. It is a chemically stable polymer with a melting point of 61° and it also exhibits a low viscosity in the molten state which allows it to be used as a carrier for the preparation of solid dispersion by fusion method [4]. It enhances solubility by reducing particle aggregation, eliminating crystallinity, increasing wettability and dispersibility, and altering the surface properties of drug particles [5]. PVP K30 is a hydrophilic polymer that has also been used successfully for the preparation of solid dispersion systems.
In order to prepare solid dispersions, solvent or fusion method is commonly adopted. Each method has some advantages and limitations. In the fusion method, it can be ensured that a solid solution is formed as a certain fraction of the drug may remain molecularly dispersed, depending on its solubility in the carrier used. Fusion method is useful in the absence of a common solvent. However, risk of drug-matrix incompatibility, phase separation and risk of drug degradation during production may be encountered in this method. On the other hand, solvent method can be used for thermolabile drugs, as minimal heat is required in the process and solid dispersions prepared by this method are amorphous in nature which improves solubility. However, the solvent chosen should be nontoxic in nature with optimum polarity in order to solubilize the drug and the polymer. Similar to the fusion method, use of solvent method also poses a risk of phase separation [6,7].
The nature of carriers used to prepare solid dispersions typically influence the type of method employed. However, in some situations it is possible to explore both methods of solid dispersion preparation. In order to minimize unnecessary experiments it would be appropriate to identify if either of the two methods offer significant advantages in terms of drug dissolution. In literature several research papers have been reported in the preparation of solid dispersions using the solvent and fusion methods [8‑12].
Efavirenz (EFV) is an antihuman immunodeficiency virus (antiHIV) drug that works by inhibiting the non‑nucleoside reverse transcriptase of HIV and is used as a part of the highly active antiretroviral therapy. EFV is freely soluble in methanol, but it is practically insoluble in water (4 µg/ml) and has a bioavailability of 40 to 45%, which makes it a suitable candidate for solid dispersion formulation [13,14].
In the present study, solid dispersions of EFV were prepared using fusion and solvent method with polyethylene glycol 8000 (PEG 8000), polyvinylpyrrolidone K30 (PVP K30) and an equal combination of both to form the binary solid dispersion systems, at drug to carrier ratios of 1:5, 1:10, and 1:15 as these excipient ratios have shown strong enhancing power on the dissolution of several drugs [15,16]. The nonionic surfactant, Tween 80 was used as the third component in the ternary system in an attempt to further enhance the dissolution rate of EFV [17]. In order to characterize the physicochemical properties of the solid dispersions, the formulations were tested using differential scanning calorimetry (DSC), powder X‑ray diffraction, Fourier transform infrared (FT‑IR) spectroscopy, and scanning electron microscopy (SEM).
Materials and Methods
EFV was procured from Aurobindo Pharma, Hyderabad, India. The polymers and surfactant (PVP K30, PEG8000 and Tween 80) were purchased from Sigma–Aldrich (M) Sdn, Petaling Jaya, Malaysia. All other materials and reagents were of analytical grade.
Preparation of solid dispersions
EFV solid dispersions of both binary and ternary systems were prepared by solvent method (SM) and fusion method (FM). In the binary system the required weights of EFV, PEG 8000 and/or PVP K30 were taken in three different drug‑polymer weight ratios (1:5, 1:10, and 1:15). In the ternary system, Tween 80 (T80) was incorporated into all the 1:10 formulations by adding 10% of the total weight to obtain drug:polymer:surfactant weight ratio of 1:10:1.1. The composition for each of the formulations is shown in Table 1.
| Formulations | Ratios | Ingredients |
|---|---|---|
| SM PEG | 1:5 | EFV:PEG 8000 |
| SM PEG | 1:10 | EFV:PEG 8000 |
| SM PEG | 1:15 | EFV:PEG 8000 |
| SM PEGT80 | 1:10:1.1 | EFV:PEG 8000:Tween 80 |
| SM PVP | 1:5 | EFV:PVP K30 |
| SM PVP | 1:10 | EFV:PVP K30 |
| SM PVP | 1:15 | EFV:PVP K30 |
| SM PVPT80 | 1:10:1.1 | EFV:PVP K30: Tween 80 |
| FM PEG | 1:5 | EFV:PEG 8000 |
| FM PEG | 1:10 | EFV:PEG 8000 |
| FM PEG | 1:15 | EFV:PEG 8000 |
| FM PEGT80 | 1:10:1.1 | EFV:PEG 8000:Tween 80 |
| FM PEGPVP | 1:5 | EFV:PEG 8000 and PVP K30 |
| FM PEGPVP | 1:10 | EFV:PEG 8000 and PVP K30 |
| FM PEGPVP | 1:15 | EFV:PEG 8000 and PVP K30 |
| FM PEGPVPT80 | 1:10:1.1 | EFV:PEG 8000 and PVP K30:Tween 80 |
PVP=Polyvinylpyrrolidone, PEG=Polyethylene glycols, EFV=Efavirenz
Table 1: Formulation table for the solid dispersions.
Fusion method
In the fusion method, the appropriate amount of polymer was melted at a temperature of 80±1°. EFV was dissolved in the molten polymer by constant stirring for 15 min and cooled rapidly in an ice bath for 2 h. This mixture was kept in a refrigerator at 4° for 3 days to solidify. All the resulting solid dispersions were scraped, pulverized in a mortar and sieved through a 45 mesh sieve. The solid dispersions were then stored in an amber glass vials and kept in the dessicator at 20±1° until further analysis.
Solvent method
In the solvent method minimal amount of methanol was used to dissolve EFV and the polymers by continuous stirring with a magnetic stirrer for an hour at room temperature. In order to completely dissolve PEG 8000, the samples were heated to 40°. Methanol was removed under reduced pressure using a rotary evaporator (Model R‑215, Buchi, Switzerland) kept at 40° until all the solvents were evaporated. The solid dispersions formed were further dried in an oven at 40° for 24 h. All the resulting solid dispersions were scraped, pulverized in a mortar and sieved through a 45 mesh sieve. Following that, all solid dispersions were stored in amber glass vials and kept in the dessicator at 20±1° until further analysis.
High performance liquid chromatography analysis
High performance liquid chromatography (HPLC) was performed using Shimadzu equipment consisting of a CBM 20A system controller, LC‑20AD pump, a DGU‑20A online degasser, a SK‑20A5C autosampler, and a CTO‑20AC column oven. Data were acquired and processed with LC software.
The analytical column used was Phenomenex Prodigy 5u, ODS 3 1OOA, 150×4.6 mm. The sample injection volume was 20 µl and UV detection was performed at 247 nm. Chromatographic analyses were performed at 30°, with a flow rate of 1 ml/min in a 15 min run time by gradient elution. The column was equilibrated for 30 min prior to the injection of the drug solution. The retention time for EFV is 6.5 min.
The mobile phase comprised phosphate buffer solution of pH 6.0 and acetonitrile (44:56 v/v). The phosphate buffer was prepared by mixing 1:1 ratio of monobasic potassium phosphate buffer and dibasic sodium phosphate buffer. Acetonitrile was used as diluent. The mobile phase was filtered through 0.45 µm membrane filter and degassed in ultrasonic bath for 30 min.
Dissolution studies
Samples of solid dispersions equivalent to 5 mg of EFV were added to 900 ml of 0.5% (w/v) SLS in water and maintained at 37±0.5°. Paddles were rotated at 100 rpm. Two microliter of aliquots at intervals of 5, 10, 15, 30, 45, 60, 90, and 120 min were withdrawn and filtered through 0.45 µm pore size filters. The same volume of fresh dissolution medium kept at 37° was substituted. Each sample was analyzed using HPLC with UV detection at 247 nm. Each dissolution test was carried out in triplicate.
Differential scanning calorimetric analysis
DSC thermograms of EFV, PEG8000, PVP K30 and all the 1:10 formulations of drug‑polymer weight ratio were recorded on DSC Q1000 (TA Instruments, New Castle, DE). Samples (7 mg weighed to a precision of 0.1 mg) were placed in aluminum pans and the lids were crimped using a TA crimper. Thermal behavior of the samples was investigated at a scanning rate of 10°/min, covering a temperature range of 25-200° against an empty aluminum pan as reference. The instrument was calibrated with an indium standard.
X‑ray powder diffraction studies
X‑Ray powder diffraction (XRPD) studies were carried out using an automated X‑ray diffractometer, Brucker Model D8 Advance (Bruker AXS, Karlsruhe, Germany). The selected samples were loaded into a specimen holder ring and held in a place on a quartz plate for exposure to Cu K‑α radiation of wavelength 1.5406 Å. The diffractometer was operated at 40 kV, 40 mA over a 2θ range of 2-60°, with a step size of 0.02°, and a count time of 1 s/step.
Fourier transform infrared spectroscopy
FT‑IR was performed on a Bruker Tensor 37 Spectrometer (Bruker Optics, Ettlingen, Germany). Samples analyzed over a scanning range of 600-4000 cm−1 were at a resolution of 4 cm−1
Scanning electron microscopy
Electron micrographs of samples were obtained using a scanning electron microscope Philips XL30S FEG (The Netherlands, LEO 440i, UK) operating at 5 kV. The specimens were mounted on a metal stub with double sided adhesive tape and coated with platinum under vacuum at 5-10 mA, 1.1 kV.
Data analysis
Statistical analysis of the dissolution parameters was carried out using the two‑way analysis of variance (ANOVA). Significance was tested at the 0.05 level of probability. Statistical analysis was performed with the software package SPSS®.
Results and Discussion
Dissolution data were evaluated on the basis of cumulative percentage drug release, which was plotted against time. Fig. 1a shows the dissolution profile of EFV and EFV solid dispersions in PEG 8000 prepared by the fusion method. All the solid dispersions prepared in varying PEG 8000 ratios showed an improved drug release compared to pure EFV sample. All the solid dispersion systems showed rapid drug release (40-60%) in the first 10 min followed by a gradual release over 2 h. EFV solid dispersion in PEG 8000 prepared in a 1:5 ratio showed a slightly reduced drug release compared to formulations that were prepared with the increased ratio of PEG 8000. The solid dispersion formulation with PEG 8000 and Tween 80 showed a similar release profile to solid dispersion without Tween 80. Similar findings were also observed in solid dispersions prepared with an equal combination of PEG and PVP (fig. 1b).

Fig. 1: Dissolution profiles of drug and solid dispersions formed by the fusion method.
Fig. 2a shows the dissolution of EFV and solid dispersions of EFV in PEG 8000 prepared by the solvent method. All the solid dispersion systems showed a 75-100% EFV release compared to a maximum release of around 20% from the pure EFV sample. Addition of Tween 80 to PEG 8000 prepared by the solvent method showed a further improvement in the dissolution parameters. A similar release pattern was observed with solid dispersion in PVP K30 prepared by the solvent method (fig. 2b). However, solid dispersions in PVP K30 prepared in a 1:5 ratio showed 80% drug release in 30 min.

Fig. 2: Dissolution profiles of drug and solid dispersions by solvent method.
It was observed that an increase in the ratio of PEG 8000 improved drug release. Hence, a 1:10 ratio of drug and the polymer were selected to compare the effectiveness of the fusion and solvent method. Fig. 3 compares the dissolution profile of EFV from the solid dispersions prepared in PEG 8000 in 1:10 ratio. After 30 min, solid dispersions prepared by solvent method showed 77% EFV release compared to 97% EFV release by the fusion method. However, after 2 h 100% drug release was obtained by the fusion method as against 95% release from the solvent method. Statistical analysis showed that there was no significant difference in the % EFV release from solid dispersions prepared by the fusion and solvent method.

Fig. 3: Dissolution profiles of drug and solid dispersions formed with PEG 8000.
For better comparison of the formulations, the dissolution data up to 10 and 30 min; Q10 and Q30 (i.e., percentage of drug release in 10 and 30 min, respectively) are calculated in Table 2. The corresponding dissolution efficiencies are shown in Table 2. Dissolution efficiency was proposed by Khan and Rhode and is defined in Eqn.1 [18]. It is the area under the dissolution curve obtained by trapezoidal rule between time points of t1 and t2, expressed as a percentage of the curve at maximum dissolution, y100, over the same time period. Dissolution efficiency (%)
| Formulation | Dissolution parametersa | |||
|---|---|---|---|---|
| *Q10 | *Q30 | **% DE10 | **% DE30 | |
| Drug (EFV) | 1.26±0.20 | 5.57±0.25 | 0.32±0.05 | 2.39±0.03 |
| FM PEG 1:10b | 60.45±7.97 | 97.26±1.21 | 17.13±2.24 | 59.69±2.09 |
| FM PEG T80 | 39.95±1.55 | 89.92±2.23 | 12.44±0.26 | 53.76±0.89 |
| FM PEGPVP 1:10 | 57.53±2.13 | 85.62±0.52 | 17.13±0.08 | 55.54±1.02 |
| FM PEGPVP T80 | 58.85±4.34 | 84.21±2.35 | 24.26±0.12 | 58.02±1.28 |
| SM PEG 1:10c | 35.52±1.84 | 76.74±3.40 | 9.08±0.54 | 47.32±2.15 |
| SM PEG T80 | 64.80±5.48 | 76.62±6.94 | 20.88±1.55 | 56.35±1.50 |
| SM PVP 1:10 | 72.08±2.49 | 90.21±0.97 | 25.29±1.73 | 66.76±0.01 |
| SM PVP T80 | 73.43±1.890 | 93.31±2.82 | 26.64±0.95 | 65.19±1.08 |
aMean±standard deviation, n=3; bFM=Solid dispersions prepared by fusion method, and cSM=Solid dispersions prepared by the solvent method, *Q10 and Q30=Percent drug dissolved in 10 and 30 min, **% DE10 and % DE30=Dissolution efficiency at t=10 min and t=30 min, PVP=Polyvinylpyrrolidone, PEG= Polyethylene glycols, EFV: Efavirenz
Table 2:in vitro dissolution data of efavirenz and the solid dispersions.
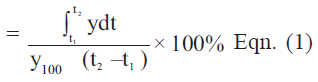
As shown in the data, the dissolution rates of all the solid dispersions were remarkably faster than the pure drug (P<0.05). Solid dispersions formed using PVP K30 alone showed the best dissolution profile as compared to formulations formed using PEG 8000 or the combination of PEG 8000 and PVP K30. Only the solid dispersions are formed using the solvent method showed further improvement in dissolution with the addition of Tween 80. From the data shown in Table 2, it is observed that EFV‑PVP K30 solid dispersions exhibited a higher percentage of drug release compared to EFV‑PEG 8000 and EFV‑PEGPVP formulations. The DE10 and DE30 for EFV‑PVP K30 solid dispersion were observed to be as high as 25.29 and 66.76%, respectively.
The thermograms for pure EFV, PEG 8000, and the solid dispersions of all the 1:10 formulations are shown in fig. 4. EFV exhibited a single, sharp endothermic peak corresponding to the melting of the drug at 138.21° with the enthalpy of fusion (ΔH) of 54.56J/g, indicating its crystalline nature [19]. The thermograms of PEG 8000 and PVP K30 showed melting endotherms at 62.29° and 114.92° with ΔH of 174.1 and 256.2 J/g, respectively. Formulations containing PEG 8000 showed that the ΔH for PEG 8000 was reduced to a range of 147.1 to 165.4 J/g. The ΔH for PVP K30 of the solid dispersions with PVP was reduced as well, to a range of 143.7 to 171.7 J/g. The ΔH for PEG 8000‑PVP K30 of the solid dispersions with both PEG 8000 and PVP K30 were reduced to even greater extent ranging from only 70.99 to 81.49J/g.

Fig. 4: DSC thermograms.
Powder X‑ray diffractograms of EFV, PEG 8000, PVP K30, and their 1:10 solid dispersions are shown in fig. 5. Major characteristic diffraction peaks of EFV are observed at 2θ diffraction angle of 6.12, 10.48, 11.00, 12.32, 13.30, 14.22, 16.96, 19.26, 20.18, 21.30, and 24.96. XRD spectrum of PEG 8000 showed two prominent peaks with a high intensity at 2θ diffraction angle of 19.12 and 23.34. The diffraction patterns of all the samples of 1:10 solid dispersions showed peaks due to PEG 8000 or patterns similar to PVP K30. Major diffraction peaks corresponding to EFV were absent and no new peaks were observed in the formulations.

Fig. 5: X‑ray diffraction patterns.
FT-IR was used to characterize possible interactions between the drug (EFV) and the polymeric carrier (PEG 8000 and PVP K30) in the solid state. Fig. 6 shows the comparison of the spectrum of pure EFV and the polymers with all the solid dispersions in a ratio of 1:10 formed by the fusion method and solvent method.

Fig. 6: FTIR spectra.
The standard spectrum of EFV shows characteristic absorption of C=O (carbonyl) in the cyclic carbonate group with a high intensity peak at 1749 cm−1. The drug also exhibits absorption at 2251 and 3317 cm−1 indicating the exocyclic tricyclic triple bond and the stretching of N‑H, respectively [20]. Important vibrations detected in the spectrum of PEG 8000 were the C‑H stretching at 2880 cm−1 and the C‑O (ether) stretching at 1110 cm−1 [21,22]. The spectrum of PVP K30 showed important bands at 1652 cm−1 which indicates C=O stretch in the cyclic amide and a broad band at about 2850-3000 cm−1 attributed to aliphatic C‑H stretch. The broad band visible at about 3000-3700 cm‑1 in PVP K30 spectrum is associated with O‑H stretching of absorbed water [23,24] confirming the broad endotherm detected in DSC.
As morphology of drug particles has an impact on micromeritic properties and dissolution behavior, the morphology of solid dispersion samples was investigated using SEM. Fig. 7 reveals the surface topography studies performed using scanning electron microscope on pure EFV together with the solid dispersions of both fusion and solvent methods. EFV powder has appeared as smooth‑surfaced, rectangular crystals in shape. All the solid dispersions show similar morphology regardless of the different methods and ratios of drug‑polymer used. The solid dispersions were observed as irregular shaped agglomerates of the drug in the polymer matrix that appeared in the form of smooth, uniform, and homogeneously mixed mass.

Fig. 7: Scanning electron micrographs.
United States Food and Drug Administration recommends 2% sodium lauryl sulphate (SLS) for EFV tablet and 1% SLS for EFV capsule as a dissolution media. However, in this study the dissolution tests were carried out in deionized water containing 0.15% w/v SLS as any concentration above 0.15% showed an undesirable dissolution profile of the solid dispersions and 100% release was observed in less than 5 min which was far too rapid. Also, 0.15% SLS was the most appropriate dissolution medium in which a distinctive difference in dissolution profile between the formulations and the pure drug could be seen, allowing a comprehensive comparison between them. From the dissolution study it was observed that only 26% of the drug was released after 2 h from the pure EFV sample. In comparison to this the dissolution rates of all the solid dispersions were remarkably enhanced (P<0.05). This increase in drug release rate from solid dispersions can be due to several reasons. Reduction of drug crystal size, absence of drug aggregation and agglomeration, conversion of drug from crystalline form to amorphous state, inhibition of crystal growth and increase in wettability by the polymers could be possible explanations for the improvement in dissolution. The polymer increases the wettability by forming a layer around the drug, thus reducing the hydrophobicity of EFV. The loss of drug crystallinity in the solid dispersion systems were confirmed by XRD, DSC, and SEM [25,26].
Increasing the proportion of polymer to drug showed an improvement in drug release over 2 h compared to pure EFV sample. This phenomenon was observed for all the polymers that were tested. However, the dissolution profile for the drug-polymer ratio of 1:15 and 1:10 did not show any marked difference in the release rate. Hence, a 1:10 drug-polymer ratio was considered as an optimum weight ratio. Akbuga et al. observed a similar finding and reported that using a 1:10 drug polymer ratio for the preparation of solid dispersion allows complete dispersion of the drug in the polymer matrix [27]. During dissolution testing it was observed that the nature of the carrier also affected dissolution. The PVP K30 formulations showed a higher dissolution efficiency compared to solid dispersions with PEG 8000 and the PEG‑PVP combination. This may be due to more wetting and solubilizing effect of PVP K30 compared to PEG 8000. Incorporation of Tween 80 showed that only the solid dispersions formed from solvent method with PEG 8000 and Tween 80 showed significant improvement in dissolution as compared to that with PEG 8000 alone.
PVP K30 has a melting point at 150° which is the temperature at which EFV is susceptible to degradation. Hence, it was not feasible to prepare EFV‑PVP K30 solid dispersion by the fusion method. However, to test the effect of PVP K30 is solid dispersions prepared by the fusion method, a combination of PEG 8000 and PVP K30 (50:50) were employed. The results did not show an improvement in the dissolution, evident from the Q10 and DE30 of FM PEGPVP 1:10 attaining only 57.53 and 55.54%, respectively, compared to 60.45 and 59.69% of FM PEG 1:10. The reason for this could be due to an insufficient amount of PEG 8000 available to lower the temperature for complete melting of PVP K30 which could have resulted in unequal distribution of drug and excipients. A slight improvement in dissolution could be seen from the 1:10 solid dispersion of EFV‑PEG 8000 prepared by the fusion method as compared to the solvent method. It was observed that EFV % release was not affected (P>0.05) by the type of method that was chosen to prepare the solid dispersion systems.
In DSC studies, the complete disappearance of the endothermic peak (a characteristic of EFV) in all the formulations could be attributed to its amorphous character in the fused state, strongly indicating that the drug is well dispersed in the polymer matrix and its recrystallization is restrained [28,29]. The amorphous form of the drug, which is the highest energy form of a compound, would be a possible explanation for the improvement in dissolution [30]. It is also reported that the deviation in peak height or the disappearance of the melting peak of the drug indicates the formation of solid dispersion [31]. The results of DSC are thus suggestive of maximal, successful complex formation in the dispersed state.
The thermal behavior of EFV in ternary systems was similar to that of the binary systems. These results indicated that Tween 80 did not play a role in the thermal behavior of EFV. The decrease in enthalpy of fusion for the polymers could be due to a decrease in PEG 8000 and PVP K30 crystallinity in the formulations, supporting the enhanced drug release that was observed in dissolution studies. Solid dispersions in PEG 8000‑PVP K30 prepared by the fusion method showed the greatest reduction in enthalpy of fusion. From this, it was expected that this solid dispersion system would have the least amount of crystallinity which would lead to an improved dissolution rate. However, the dissolution rates of the PEG 8000‑PVP K30 formulations were surprisingly similar to the other solid dispersion systems giving rise to a speculation that the ratio of PEG 8000 to PVP K30 was probably not optimized.
The XRD study was carried out to investigate the crystallinity of EFV in PEG 8000, PVP K30, and a mixture of PEG 8000 and PVP K30. The presence of numerous distinct peaks in the XRD spectrum indicates that EFV was present as a crystalline material. PEG 8000 also exhibited a distinct pattern with two diffraction peaks with the highest intensity. On the other hand, the spectrum of PVP K30 was characterized by the complete absence of any diffraction peak, which is characteristic of an amorphous compound. The incorporation of Tween 80 had no effect on XRD patterns of EFV in the solid dispersion system. It was considered that Tween 80 might exist in the amorphous region of both EFV and PEG 8000 [32]. The diffraction patterns of all the samples of 1:10 solid dispersions showed peaks due to PEG 8000 or patterns similar to PVP K30. The absence of major diffraction peaks corresponding to EFV suggests that EFV was present as an amorphous material inside the PEG 8000 or PVP K30 matrix. Hence, the increase in dissolution of the formulations could be a result of the amorphous drug. No other peaks than those that could be assigned to the mixture of PVP K30 and PEG 8000 were detected in solid dispersions, indicating the absence of chemical interaction in the solid state between the three entities. The positions of PVP K30 and PEG 8000 patterns in the solid dispersions were the same and superimposable, which again ruled out the possibility of chemical interaction between EFV, PVP K30, and PEG 8000.
From the structures of EFV, PEG 8000, and PVP K30, it can be assumed that the possible interaction would be hydrogen bonding between C=O and N‑H group of EFV with the lone pair of electrons of the oxygen atom in PEG 8000. In the case of PVP, which consists of repeating units of 1‑ethynyl‑2‑pyrrolidione monomer, there are two electron donating centers in PVP K30 (C=O group and N atom of the pyrrole ring) which are capable of forming hydrogen bonds. However, carbonyl group is more favorable due to the steric hindrance effect on the nitrogen atom [33]. Thus, any sign of interaction would be reflected by band shifts, broadening, disappearance of peaks or intensity alterations as compared to the spectra of the pure drug and polymers [34,35].
The N‑H stretch vibration region of EFV disappeared in all the solid dispersion systems and there was reduction in the peak and slight shift of C=O stretch in all of the investigated formulations. This could be due to physical interactions between the drug and the polymers suggesting intermolecular hydrogen bonding. This could also be due to the change of crystalline form of EFV to the amorphous form as confirmed by XRD. The characteristic peak of EFV exocyclic tricyclic triple bond (2251 cm−1) was absent indicating the trapping of EFV in the polymer matrix. In ternary solid dispersions, the characteristic vibration wave of Tween 80 was almost shielded by the peaks of polymers. The absence of the O‑H stretching of the terminal hydroxyl group of Tween 80 in the spectra of all the investigated solid dispersions indicated that there is intermolecular hydrogen bonding between Tween 80 and the drug or polymers [36]. In all the solid dispersions systems EFV is converted from a crystalline form into an amorphous form. The stabilizing polymers added to the solid dispersion system cover the hydrophobic surface of the precipitated crystals providing stearic hindrance, thus preventing the crystal growth [37]. The crystalline properties of EFV seemed to have diminished during the preparation of solid dispersions. Hence, the absence of crystals not only indicates good miscibility between the drug and the polymers but also the formation of an effective and successful solid dispersion system.
Acknowledgements
The authors would specially like to thank Asma Othman, Sairam Bahera, and Joyti Gupta for their assistance in the lab.
References
- Rinaki E, Valsami G, Macheras P. Quantitative biopharmaceutics classification system: The central role of dose/solubility ratio. Pharm Res 2003;20:1917‑25.
- Iqbal Z, Babar A, Ashraf M. Controlled‑release naproxen using micronized ethyl cellulose by wet‑granulation and solid‑dispersion method. Drug DevInd Pharm 2002;28:129‑34.
- Chiou WL, Riegelman S. Pharmaceutical applications of solid dispersion systems. J Pharm Sci 1971;60:1281‑302.
- Law D, Wang W, Schmitt EA, Qiu Y, Krill SL, Fort JJ. Properties of rapidly dissolving eutectic mixtures of poly (ethylene glycol) and fenofibrate: The eutectic microstructure. J Pharm Sci 2003;92:505‑15.
- Biswal S, Sahoo J, Murthy PN. Characterisation of gliclazide‑PEG 8000 solid dispersions. Trop J Pharm Res 2009;8:417‑24.
- Serajuddin AT. Solid dispersion of poorly water‑soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J Pharm Sci 1999;88:1058‑66.
- Dhirendra K, Lewis S, Udupa N, Atin K. Solid dispersions: A review. Pak J Pharm Sci 2009;22:234‑46.
- Chaulang G, Patel P, Hardikar S, Kelkar M, Bhosale A, Bhise S. Formulation and evaluation of solid dispersions of furosemide in sodium starch glycolate. Trop J Pharm Res 2009;8:43‑51.
- Dhanaraju MD, Enose Arno A, Sundarseelan, Prasanna PM, Krishnan RK. A study of solid dispersion of griseofulvin with polyethylene glycol 6000: Polysorbate 80 mixture. Indian Drugs 2001;38: 633‑637.
- Franco M, Trapani G, Latrofa A, Tullio C, ProvenzanoMr, Serra M, et al. Dissolution properties and anticonvulsant activity of phenytoin‑polyethylene glycol 6000 and polyvinylpyrrolidone K‑30 solid dispersions. Int J Pharm 2001;225:63‑73.
- Jain SK, Shukla M, Shrivastava V. Development and in vitro evaluation of ibuprofen mouth dissolving tablets using solid dispersion technique. Chem Pharm Bull 2010;58:1037‑42.
- Najmuddin M, Khan T, Mohsin AA, Shelar S, Patel V. Enhancement of dissolution rate of ketoconazole by solid dispersion technique. Int J Pharm PharmSci 2010;2:132‑6.
- Csajka C, Marzolini C, Fattinger K, Dacosterd LA, Fellay J, Telenti A, et al. Population pharmacokinetics and effects of efavirenz in patientswith human immunodeficiency virus infection. ClinPharmacolTher 2003;73:20‑30.
- Chiappetta DA, Hocht C, Taira C, Sosnik A. Efavirenz‑loaded polymeric micelles for pediatric anti‑HIV pharmacotherapy with significantly higher oral bioavailaibility. Nanomedicine 2010;5:11‑23.
- Craig DQ. Polyethylene glycols and drug release. Drug DevInd Pharm 1990;16:2501‑26.
- Van Den Mooter G, Augustijns P, Blaton N, Kinget R. Physico‑chemical characterization of solid dispersions of temazepam with polyethylene glycol 6000 and PVP K30. Int J Pharm 1998;164:67‑80.
- Okonogi S, Puttipipatkhachorn S. Dissolution improvement of high drug‑loaded solid dispersion. AAPS Pharm Sci Tech 2006;7:E148‑53.
- Khan CA, Rhodes CT. The concept of dissolution efficiency. J Pharm Pharmacol 1975;27:48‑9.
- Shah J, Vasanti S, Anroop B, Vyas H. Enhancement of dissolution rate of valdecoxib by solid dispersions technique with PVP K 30 and PEG 4000: Preparation and in vitro evaluation. J InclPhenomMacrocyclChem 2009;63:69‑75.
- Indrajit S, Banerjee S, Ramchandran AV, Geckeler KE, Murthy CN. Synthesis of cyclodextrin and sugar‑based oligomers for the efavirenz drug delivery. MacromolSymp 2010;287:51‑9.
- Khattab IS, Nada A, Zaghloul AA. Physicochemical characterization of gliclazidemacrogol solid dispersion and tablets based on optimized dispersion. Drug DevInd Pharm 2010;36:893‑902.
- Patel RP, Patel DJ, Bhimani DB, Patel JK. Physicochemical characterization and dissolution study of solid dispersions of furosemide with polyethylene glycol 6000 and polyvinylpyrrolidone k30. DissolutTechnol 2008;15:17‑25.
- Patel RP, Patel MM. Physicochemical characterization and dissolution study of solid dispersions of lovastatin with polyethylene glycol 4000 and polyvinylpyrrolidone K30. Pharm Dev Tech 2007;12:21‑33.
- Sethia S, Squillante E. Solid dispersion of carbamazepine in PVP K30 by conventional solvent evaporation and supercritical methods. Int J Pharm 2004;272:1‑10.
- Mura P, Moyano JR, González‑Rodríguez ML, Rabasco‑Alvaréz AM, Cirri M, Maestrelli F. Characterization and dissolution properties of ketoprofen in binary and ternary solid dispersions with polyethylene glycol and surfactants. Drug DevInd Pharm 2005;31:425‑34.
- Ford JL. The current status of solid dispersions. Pharm ActaHelv 1986;61:69‑88.
- Akbuga J, Gursoy A, Kendi E. The preparation and stability of fast release furosemide‑PVP solid dispersion. Drug DevInd Pharm 1988;14:1439‑64.
- Park YJ, Ryu DS, Li DX, Quan QZ, Oh DH, Kim JO, et al. Physicochemical characterization of tacrolimus‑loaded solid dispersion with sodium carboxylmethyl cellulose and sodium lauryl sulfate. Arch Pharm Res 2009;32:893‑8.
- Joe JH, Lee WM, Park YJ, Joe KH, Oh DH, SeoYG, et al. Effect of the solid‑dispersion method on the solubility and crystalline property of tacrolimus. Int J Pharm 2010;395:161‑6.
- Rama Rao N, Chowdary KP. Improvement of dissolution rate and bioavailability of piroxicam with pregelatinized starch. Indian J Pharm Sci 2001;63:36‑40.
- Rawat S, Jain SK. Solubility enhancement of celecoxib using β‑cyclodextrin inclusion complexes. Eur J Pharm Biopharm 2004;57:263‑7.
- Morris KR, Knipp GT, Serajuddin AT. Structural properties of polyethylene glycol‑polysorbate 80 mixture, a solid dispersion vehicle. J Pharm Sci 1992;81:1185‑8.
- Taylor LS, Zografi G. Spectroscopic characterization of interactions between PVP and indomethacin in amorphous molecular dispersions. Pharm Res 1997;14:1691‑8.
- MartínezOhárriz MC, Rodríguez‑Espinosa C, Martín C, Goñi MM, Trosilarduya MC, Sánchez M. Solid dispersions of diflunisal‑PVP: Polymorphic and amorphous states of the drug.
- El‑Hinnawi MA, Najib NM. Ibuprofen‑polyvinylpyrrolidone dispersions. Proton nuclear magnetic resonance and infrared studies. Int J Pharm 1987;37:175‑7.
- Zheng Y, Eli W, Li G. FTIR study of tween80/1-butyl‑3-methylimidazolium hexafluorophosphate/toluene microemulsions. Colloid PolymSci 2009;287:871‑6.
- Talari R, Varshosaz J, Mostafavi SA, Nokhodchi A. Dissolution enhancement of gliclazide using pH change approach in presence oftwelve stabilizers with various physico‑chemical properties. J Pharm PharmSci 2009;12:250‑65.