- *Corresponding Author:
- N. A. Al-shar’i
Department of Medicinal Chemistry and Pharmacognosy, Faculty of Pharmacy, Jordan University of Science and Technology, P.O. Box 3030, Irbid 22110, Jordan
E-mail: nashari@just.edu.jo
| Date of Received | 25 November 2019 |
| Date of Revision | 02 April 2020 |
| Date of Acceptance | 02 May 2020 |
| Indian J Pharm Sci 2020;82(3):472-482 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
The checkpoint kinase, CHK1 plays an important role in repairing DNA damage induced by genotoxic agents for cancer treatment, which renders the cells resistant to such treatment modalities. Therefore, its inhibition appeared as a highly attractive strategy to re-sensitize cancer cells to the DNA damaging chemo and radiotherapies. In this study, a structure-based drug design approach was employed to identify potential ATP competitive CHK1 inhibitors. To this end, 3 crystal structures of CHK1 in complex with ATP competitive inhibitors were utilized to generate structure-based pharmacophore models that were subsequently used for virtual screening of commercial databases. Retrieved hits were filtered, then they were docked into the binding site of the enzyme and the free binding energies of the top-ranked docked hits were calculated. Based on the in silico results, 10 compounds were selected and their CHK1 inhibitory potential was evaluated in vitro. Noteworthy, in this exploratory study compounds 7 and 9 showed moderate but significant inhibition of the catalytic activity of the CHK1 enzyme. These two compounds were identified as promising hits worthy of further optimization towards designing better leads with drug like properties as potential competitive CHK1 inhibitors.
Keywords
Serine-threonine checkpoint kinase 1, CHK1, pharmacophore modelling, virtual screening, molecular docking
Among the many feasible targets available to develop anticancer therapies, kinases appear to be one of the most attractive, due to the prominent role played by the phosphorylation process in regulating almost every cellular aspect, and the role of their overexpression and dysregulation in cancer development and metastasis[1,2].
The checkpoint kinase, CHK1, which belongs to the serine/threonine family of kinases, has a major role in regulating the G2/M phases of the cell cycle. Under normal conditions, DNA damage, which is brought about either by endogenous or by environmental insults, is sensed by sensor multiprotein complexes, which activate the proximal transducers; ataxia telangiectasia mutated and ataxia telangiectasia and Rad3-related that subsequently induces phosphorylation and activation of their distal transducer, CHK1. The activated CHK1 then affects the activation of a number of downstream effectors, eventually directing the activation of different repair mechanisms[3].
When cancer cells are exposed to genotoxic chemo- or radiotherapies, they respond by overexpressing CHK1, which facilitates DNA damage repair and render them resistant to treatment. Should CHK1 be inhibited, cancer cells will lose the ability to repair DNA damage. Consequently, CHK1 had appeared as potential target for therapeutic intervention, augmented by the fact that up to 70 % of cancer cells rely solely on CHK1 for damage repair, while normal cells have repair mechanisms other than CHK1. Hence, targeting CHK1 would allow the development of selective inhibitors against cancer cells while sparing normal cells from inhibitory effects[4,5]. Based on these facts, several CHK1 inhibitors have been identified[6,7], however, up to now none of the developed inhibitors had succeeded to be further evolved into a drug[7]. Therefore, due to its critical role in enhancing the efficacy of DNA damaging chemotherapies, further drug design efforts are needed to develop CHK1 inhibitors. Herein, a structurebased drug design approach was employed toward the development of new ATP competitive CHK1 inhibitors.
Materials and Methods
All modelling calculations were carried out using Discovery Studio (DS) 2017 from BIOVIA Software Inc.[8] and Pipeline Pilot 2017 from Biovia Software Inc.[9]. Presentation quality images were created using DS and ChemDraw© Ultra 12.0.2. Selected compounds were purchased from Ambinter (Greenpharma), France and Maybridge Chemical Holdings Ltd., (Thermo Fisher Scientific), UK through local vendors.
Preparation of the CHK1 enzyme:
The protein databank archive contains 133 entries for CHK1. Except for the crystal structure of the apo form of the enzyme, in addition to 3 crystal structures of CHK1 in complex with allosteric inhibitors, all other crystal structures represented CHK1 complexed with ATP competitive inhibitors. Of these structures, 21 were selected for the structure-based design of ATP competitive inhibitors. The selection of those crystal structures was based on crystal resolution that need to be less than 2.5 Å; being unmutated (wild type), and complexed with ligands whose IC50 values are less than 10 nM. Only 3 out of the 21 selected crystal structures had succeeded to generate good queries that were subsequently utilized for virtual screening. The final selected crystal structures have PDB codes of 2HY0, which is complexed with indolyl quinolinone inhibitor[10]; 3PA5, complexed with a thiophene carboxamide inhibitor[11] and 3OT8, complexed with a pyrazolopyrimidine inhibitor[12]. The resolutions of these crystal structures were 1.7, 1.7, and 1.65 Å, respectively. The prepare protein protocol[13] was utilized to prepare all crystal structures using default parameters except for keeping all water molecules, which was set to true. Glycerol was deleted wherever relevant. The prepare protein protocol cleans common problems in the input protein structure where it standardize atom names, insert missing atoms in residues, remove alternate conformations, insert missing loop regions, correct connectivity and bond order, and protonate the protein at a pH of 7.4 in preparation for further processing by other protocols.
Pharmacophore modelling:
The receptor-ligand pharmacophore generation (RLPG) protocol was utilized to generate 3D pharmacophore models from the prepared crystal structures. The generated pharmacophore models were automatically validated against a test set of 19 active and 17 inactive ligands[14,15] using the parameters under the validation group within the RLPG protocol.
Virtual screening:
Three of the generated hypotheses were selected and utilized to virtually screen 2 small molecule databases; Aldrich MyriaScreen Diversity Library II and Maybridge Screening Collection, collectively containing more than 60 000 compounds. The search 3D database protocol was used for virtual screening using default parameters except for the search method which was set to BEST. Using a customized pipeline pilot protocol, retrieved hits were filtered by removing duplicates, consideration of their fit values where a threshold of 2.5 or more was set and their compliance with Lipinski and Veber rules for drug-likeness. Filtered hits were then prepared and subjected to molecular docking.
Molecular docking and calculation of binding energies:
Out of the 3 crystal structures utilized in this study, the 2HY0 crystal, which has the lowest number of missing residues, was used for molecular docking of the filtered hits. Prior to docking, the 2HY0 crystal complex was typed with CHARMm force field, solvated and minimized.
Solvation and minimization:
Using the solvation protocol, the prepared complex was solvated by immersing it in a truncated octahedral cell of explicit water with a 5 Å minimum distance from cell boundary under periodic boundary conditions. Then, the system was neutralized by adding NaCl counterions. Finally, the solvated system was minimized using the smart minimizer within the minimization protocol. The minimization process was conducted in three stages, each of 5000 steps, in order to relax the system and remove any potential clashes between the protein and the solvent. In all minimization stages the smart minimizer algorithm was set to perform 1000 steps of steepest descent, followed by conjugate gradient minimization with a root mean square (RMS) average gradient of 0.1 Kcal/(mol×Å). In the first minimization stage, only water molecules, counterions, and hydrogen atoms were minimized; in the second stage, all atoms except the backbone heavy atoms were free to relax; and in the third stage, the entire system was allowed to move. In all stages, Particle Mech Ewald[16] was utilized to treat electrostatic interactions.
Molecular docking:
All water molecules and counter ions of the minimized solvated complex were removed. Before deleting the co-crystallized ligand, it was used to define the binding site of the enzyme by a sphere of 14 Å radius using the Define and Edit Binding Site tool within DS. Furthermore, prior to ligand docking, the filtered hits were prepared using the prepare ligands protocol in which all default parameters were retained except for the ionization pH which was set to 7.2-7.6, isomers generation was set to false, and the maximum number of tautomers to generate was set to 3. The prepared hits were then docked into the defined ATP binding pocket utilizing the dock ligands (CDOCKER) protocol[17] using default parameters. All docked poses, which were scored according to their -CDOCKER energy, were rescored using LigScore1[18] and PMF04[19] scoring functions and their consensus scores were calculated.
Calculation of binding energies:
All hits that got a consensus score of 3 were in situ minimized utilizing the in situ ligand minimization protocol using default parameters. Then, their binding free energies were calculated using the Calculate Binding Energies protocol. To account for the solvent effect, the Poisson Boltzmann with non-polar surface area solvent model[20] was used. The ligand conformational entropy value was set to true, and the BEST conformation generation method was applied.
In vitro enzyme assay:
The in vitro biological activity of the selected compounds against the CHK1 enzyme was performed at Reaction Biology Corp. (Malvern, PA, USA) using recombinant human CHK1 kinase. Briefly, a substrate (peptide KKKVSRSGLYRSPSMPENLNRPR) solution was prepared in fresh assay buffer. The buffer comprised of 20 mM Hepes (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02 % Brij35, 0.02 mg/ml BSA, 0.1 mM Na3VO4, 2 mM DTT, and 1 % DMSO. The CHK1 enzyme was then delivered to the substrate solution with gentle mixing. The tested compounds were dissolved in DMSO and delivered into the reaction mixture by acoustic technology (Echo550; nanoliter range) and incubated at room temperature for 20 min. [γ-33P]-ATP was then delivered to the reaction mixture to initiate the reaction and the mixture was incubated at room temperature for 2 h. The kinase activity was detected by P81 filter binding method[21]. Staurosporine was used as a positive control in all assay experiments. A schematic representation of the entire methods applied in this study is shown in fig. 1.

Fig. 1: A flowchart summarizing the entire methodology employed in this work
Results and Discussion
Within the protein data bank, 133 crystal structures are available for CHK1. All crystal structures other than the structure that represents the apo form of the enzyme, and the 3 crystal structures for CHK1 in complex with allosteric inhibitors, were inspected to select models for the design of ATP competitive inhibitors. Initially, all crystal structures whose resolution is better than 2.5 Å, unmutated, and complexed with ligands whose IC50s are less than 1 nM, were considered. The crystal structures whose PDB codes are 2HOG[22], 2HY0[10], 2YEX[23] and 3TKI[24] fulfilled this criteria and were downloaded from the protein data bank and utilized for pharmacophores generation. However, only the 2HY0 crystal complex resulted in the generation of hypotheses that succeeded in retaining a good number of hits upon virtual screening of the databases. In the second trial, all crystal structures whose resolution is better than 2.5 Å, unmutated, and complexed with ligands with IC50 values between 1 and 5 nM were considered. The corresponding PDB codes for those structures are 2R0U[25], 2YDJ[26], 3OT3[27], 3PA3[11], 3PA4[11], 3PA5[11], 4HYH[28], 4HYI[28], 4QYG[15], 4QYH[15], 4RVM[14], 5DLS[29] and 5F4N[30]. The best-generated pharmacophore that was selected corresponds to the 3PA5 crystal structure. Finally, the crystal structures for CHK1 in complex with ligands whose IC50s were less than 10 nM, unmutated and with a resolution better than 2.5 Å were considered. The considered crystal structures have PDB codes of 2E9N[31], 2YDK[26], 3OT8[12] and 6FCF[32]. Out of those, only the 3OT8 crystal complex generated successful queries. Consequently, the final selected crystal structures were 2HY0, 3PA5, and 3OT8, which are complexed with highly potent ligands of diverse chemotypes. The 2HY0 crystal structure corresponds to CHK1 in complex with indolyl quinolinone inhibitor (compound 306) whose IC50 is 0.65 nM (fig. 2a); the 3PA5 crystal structure corresponds to CHK1 complexed with a thiophene carboxamide inhibitor (compound C73) with an IC50 of 2 nM (fig. 2b) and the 3OT8 crystal corresponds to CHK1 in complex with a pyrazolopyrimidine inhibitor (compound MI5) with an IC50 of 9 nM (fig. 2c). The resolutions for the selected crystal structures are 1.7, 1.7, and 1.65 Å, respectively.

Fig. 2: Structures of the co-crystallized ligands
Chemical structures of the co-crystallized ligands in the selected crystal complexes, A. compound 306, B. compound C73 and C. compound MI5
The 3D coordinates of the selected crystal structures were retrieved from the protein databank, then the Protein Report tool was utilized to assess their quality. The protein reports indicated that the 2HY0 structure has 7 missing residues (Arg44, Ala45, Val46, Asp47, Cys48, Pro49, and Glu50), while both the 3PA5 and the 3OT8 structures have 14 missing residues, the former is missing the residues Asp41, Met42, Lys43, Arg44, Ala45, Val46, Asp47, Cys48, Pro49, Glu50, Arg75, Glu76, Gly77, and Asn78; while the latter is missing the residues Ala19, Tyr20, Lys43, Arg44, Ala45, Val46, Asp47, Cys48, Pro49, Glu50, Glu76, Gly77, Asn78, and Ilu79. The Prepare Protein protocol was utilized to prepare the crystal structures as described in the methods section.
An important region within the CHK1 ATP binding site that is unique to this enzyme is a buried pocket lined by the amino acid residues Gly55, Asn59 and Val68 (fig. 3). Three water molecules are conserved within this pocket, which aid in ligand potency and selectivity should the ligand bind them[22,33-36]. Consequently, those water molecules were retained in all prepared structures.

Fig. 3: Crystal structure of CHK1 in complex with a competitive ATP with a close-up view
A. The crystal structure of CHK1 in complex with a competitive ATP inhibitor (PDB code 2HY0), the protein is depicted in white cartoon, ATP binding site is shown as surface colored by ionization potential, the ligand is shown in sticks with carbons colored green and the crucial water molecules are shown as red spheres. B. A close-up view showing the buried pocket and the 3 water molecules
The receptor ligand pharmacophore generation protocol, which generates a set of pharmacophore models based on the features involved in receptor-ligand interaction, was utilized to generate pharmacophore models from the three prepared complexes. The types of interactions the protocol consider are hydrogen bond donor (HBD), hydrogen bond acceptor (HBA), positive ionizable (PI), negative ionizable (NI), hydrophobic (HY), and ring aromatic (RA). All generated pharmacophores were validated against a test set gathered from reported CHK1 inhibitors in the literature. The test set included 19 active ligands with IC50 values ranging from 0.31 to 5 nM, and 17 inactive ligands with IC50 values ranging from 0.1 to >10 μM (fig. 4)[14,15].

Fig. 4: The set of ligands used in validation of the generated pharmacophore models
The output of the validation process is reported in the form of receiver operating characteristic (ROC) curves. The ROC plots represent a measure of model sensitivity (the power of identifying active compounds in the test set) and specificity (the power of identifying inactive compounds in the test set), and provides an overall quality assessment of the generated models as being good or bad based on their individual overall score. The ROC scores can take a value between 0.5 and 1.0, where a value close to 0.5 is indicative of randomness and no predictive power of the model, while a value close to 1 indicates a model having an excellent selectivity. The in between values of 0.6-0.7, 0.7-0.8, 0.8-0.9 are indicative of poor, fair, and good quality, respectively[37].
The 2HY0 crystal complex has resulted in the generation of 10 pharmacophore models, with selectivity scores ranging from 7.21 to 10.24. Having a ROC score of 0.963 and excellent quality, pharmacophore 8 (PH-1; fig. 5a) was selected to be used in virtual screening of commercial databases. This pharmacophore comprised 4 features, namely 2 HBA, one HBD and one HY, in addition to 15 exclusion spheres. The 3PA5 crystal complex has resulted in the generation of six pharmacophore models with selectivity scores ranging from 7.14 to 10.05. Based on its good quality and a ROC score of 0.848, pharmacophore 5 (PH-2; fig. 5b) was selected. This pharmacophore comprised 4 features; 2 HBA, one HY, and one PI, in addition to 18 exclusion spheres. Using the 3OT8 crystal complex has resulted in the generation of four pharmacophore models, with selectivity scores ranging from 7.35 to 8.26. Pharmacophore 1 (PH-3; fig. 5c), with a ROC score of 0.845 indicative of good quality was selected. This pharmacophore comprises four features; 2 HBD, one HBA, and one HY, in addition to 19 exclusion spheres.

Fig. 5: Selected pharmacophore models generated and their ROC curves
Upper panel- Pharmacophore models generated using the receptor-ligand pharmacophore generation protocol from each of the three crystal complexes, the feature types are HY (cyan), HBD (magenta), HBA (green), PI (red) and exclusion spheres (grey). Lower panel- ROC curves for the selected pharmacophores. A. PH-1 obtained from 2HY0 crystal complex. B. PH-2 obtained from the 3PA5 crystal complex and C. PH-3 obtained from the 3OT8 crystal complex
Using the search 3D database protocol, Maybridge and Aldrich databases were screened against the 3 generated pharmacophore models. A total of 19 345 hits were retrieved where 15 953 have been retrieved from Maybridge and 3392 from Aldrich. The retrieved hits were filtered using customized pipeline pilot protocol such that duplicate hits, hits that have a fit value less than 2.5, and hits that do not comply with Lipinski and Veber rules of drug-likeness were removed. The number of hits that passed these filtration criteria was 1324 (Table 1).
| Pharmacophore | Maybridge DB | Aldrich DB | ||
|---|---|---|---|---|
| No. of hits | Range of fit value | No. of hits | Range of fit value | |
| PH-1 | 12824 | 3.77-7.82 e-10 | 2388 | 3.70-3.40 e-9 |
| PH-2 | 1575 | 3.33-3.55 e-10 | 520 | 3.30-1.39 e-9 |
| PH-3 | 1554 | 3.15-4.25 e-8 | 484 | 3.34-3.07 e-6 |
| Subtotal number of hits | 15953 | 3392 | ||
| Total filtered hits | 1324 | |||
Table 1: Virtual Screening of Each Pharmacophore Along with Fit Value Range
Being the model with the lowest number of missing residues out of the 3 utilized crystal structures in this study, the 2HY0 crystal structure was utilized for molecular docking of the filtered hits. The structure was prepared, solvated and minimized as discussed in the methods section. Prior to docking, the binding site was defined with a sphere of 11.25 Å radius based on the co-crystalized ligand, which was increased to 14 Å to include all binding regions within the ATP binding site. All water molecules but the three conserved ones, were deleted, in addition to the co-crystallized ligand, and the counter ions.
The 1324 filtered hits were prepared as described earlier, which led to the generation of 2796 molecules. Subsequently, all prepared hits were docked into the defined binding site of the 2HY0 crystal structure using CDOCKER protocol. The CDOCKER algorithm is a grid-based molecular dynamics docking algorithm that uses CHARMm force field, and refines the docked poses via a final minimization of simulated annealing step[17].
For the purpose of checking the accuracy of the docking algorithm and the validity of the docking parameters to be used, the co-crystallized ligand was extracted from the complex and then re-docked into the binding site of the protein. The calculated RMSD of the generated poses of the docked ligand relative to the co-crystallized one reflects the accuracy of the docking algorithm. Usually a value of less than 2 Å is considered acceptable[38,39], however, a value of less than 1.5 Å is better and that of less than 1.0 is very good. The obtained RMSD was 0.41 Å, which is indicative of excellent agreement with the experimental pose.
After being validated, the protocol was then utilized to dock the prepared hits. The -CDOCKER energy scores (interaction energy plus ligand strain) of the generated poses were ranging from -110.87 to 56.23 kcal/mol. Then, all poses were rescored using the ligscore1 and PMF04 scoring functions to calculate ligand consensus scores in order to identify the compounds whose scores are within the top 20 % in all scoring functions (-CDOCKER energy, -PMF04, and LigScore1). The number of hits that got a consensus score of 3 were 27, which were then minimized in situ and their free binding energies were calculated. All 27 hits were found to bind favorably, with total free binding energies ranging from -57.35 to -14.12 kcal/mol. Based on docking scores, calculated total free binding energies, structural diversity, and visual inspection of binding poses, 10 compounds were selected for biological evaluation of their inhibitory potential against the CHK1 enzyme (Table 2). The SciFinder database was consulted for exact structure search of the selected compounds, and none were found to be previously reported as a CHK1 inhibitor.
| No. | Chemical structure | Code and database | TBEa | -CDEb | Consensus |
|---|---|---|---|---|---|
| 1 | 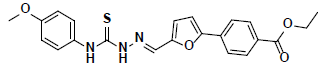 |
RDR01930 Maybridge |
-57.35 | 24.59 | 3 |
| 2 | 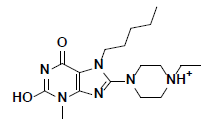 |
L217603 Aldrich |
-49.50 | 37.80 | 3 |
| 3 | 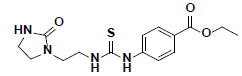 |
HTS03381 Maybridge |
-47.33 | 30.01 | 3 |
| 4 | 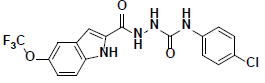 |
KM09381 Maybridge |
-44.21 | 35.93 | 3 |
| 5 | 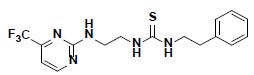 |
AW00715 Maybridge |
-42.12 | 36.62 | 3 |
| 6 | 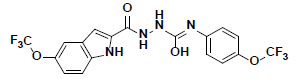 |
KM09380 Maybridge |
-41.09 | 32.25 | 3 |
| 7 | 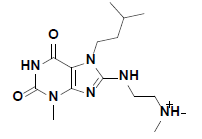 |
R143219 Aldrich |
-41.05 | 37.59 | 3 |
| 8 | 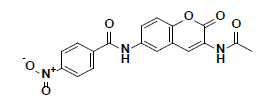 |
BTB04771 Maybridge |
-38.41 | 28.07 | 3 |
| 9 | 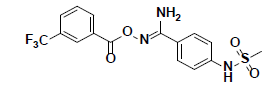 |
SPB06762 Maybridge |
-37.02 | 32.16 | 3 |
| 10 | 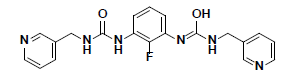 |
CD10587 Maybridge |
-34.55 | 33.35 | 3 |
aTotal Free Binding Energy (kcal/mol); bthe negative value of CDOCKER Energy (kcal/mol)
Table 2: Ten Selected Compounds for Biological Evaluation along with their Calculated Scores
The in vitro inhibitory activity of the selected compounds against the CHK1 enzyme was performed as described in the methods section. In order to establish a primary perception about the kinase inhibition profile of the 10 selected compounds, they were screened at a concentration of 10 μM and their % CHK1 inhibition was measured relative to staurosporine (the positive control). Unfortunately, the screening results did not show significant inhibitory potency at an assay concentration of 10 μM with the exception of compounds 7 and 9 which showed a modest but significant reduction in the basal activity of the kinase (approximately 12 and 20 %, respectively) (Table 3). Nevertheless, compounds 7 and 9 can be considered as hits that need further optimization in order to be converted into lead-like compounds.
| Compound No. | CHK1 residual activity (%)a | Inhibition (%) |
|---|---|---|
| 1 | 123.39 | 0 |
| 2 | 90.57 | 9.43 |
| 3 | 93.24 | 6.77 |
| 4 | 114.14 | 0 |
| 5 | 99.30 | 0.71 |
| 6 | 113.08 | 0 |
| 7 | 88.11 | 11.9 |
| 8 | 95.93 | 4.07 |
| 9 | 80.30 | 19.71 |
| 10 | 95.96 | 4.04 |
a% CHK1 inhibitory activity, average of 2 independent readings at 10 μM
Table 3: Biological Screening of the Selected Compounds at 10 mm.
The docked pose of compound 7 and its 2D interaction map are shown in fig. 6. As can be noticed from the docked pose, the compound well-occupies the ATPbinding pocket. The major types of interactions the compound forms are hydrogen bonding with Cys87 within the hinge, and with Glu91; besides numerous hydrophobic interactions with Leu15, Val23, Ala36, Tyr 86, Cys87, and Leu137. Interestingly, the protonated amine group forms an ionic interaction with Asp94 at the entrance of the ATP-binding site which is an allosteric residue. No interaction with any of the conserved water molecules was observed.

Fig. 6: Docked pose of compound 7 and the 2D interaction map
A. The top ranked docked pose of compound 7 with respect to total binding free energy score. The ATP binding pocket is shown as hydrophobic surface (blue represents hydrophilic areas and brown represents hydrophobic areas), compound 7 is shown in balls and sticks with carbons colored light green. B. A 2D interaction map showing different interactions between compound 7 and the active site of the enzyme. Residues are shown as plates and colored according to the type of their interactions with the ligand
Compound 9, whose docked pose and 2D interaction map are shown in fig. 7, also well-occupies the ATP-binding pocket. The major types of interactions the compound forms within the binding site are hydrophobic and hydrogen bonding interactions. The most important of which is the hydrogen bonding with Cys87 within the hinge region of the enzyme and a hydrophobic interaction with Leu84, the gatekeeper residue. Compound 9 also forms two hydrogen bonds with Leu15 and Ser147. Moreover, it forms numerous hydrophobic interactions with the residues Leu15, Val23, Ala36, Val68, Cys87, and Leu137. No interaction with any of the conserved water molecules was observed.

Fig. 7: Docked pose of compound 9 with 2D interaction map
A. The top ranked docked pose of compound 9 with respect to total binding free energy score and B. 2D interaction map (depictions as shown in fig. 6)
In this exploratory study, a structure-based drug design approach involving pharmacophore modelling, virtual screening, molecular docking and calculation of free binding energies was employed with the aim of identifying novel ATP competitive CHK1 inhibitors. The applied approach has resulted in the selection of 10 compounds as potential CHK1 inhibitors. The in vitro screening results, at a concentration of 10 μM of the tested compounds, showed that compounds 7 and 9 have a moderate but significant inhibition of the catalytic activity of the CHK1 enzyme. Nevertheless, the two identified compounds can be considered as hits worthy of further optimization towards designing leadlike compounds.
Acknowledgments
This work was funded by the Deanship of Scientific Research at Jordan University of Science and Technology, grant number (20190121).
References
- Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest 2015;125(5):1780-9.
- Bhullar KS, Lagarón NO, McGowan EM, Parmar I, Jha A, Hubbard BP, et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 2018;17(1):48.
- Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res 2010;16(2):376-83.
- Zhang Y, Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int J Cancer 2014;134(5):1013-23.
- Suzuki K, Matsubara H. Recent Advances in p53 Research and Cancer Treatment. J Biomed Biotechnol 2011;2011:7.
- Rundle S, Bradbury A, Drew Y, Curtin N. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers 2017;9(5):41.
- Qiu Z, Oleinick NL, Zhang J. ATR/CHK1 inhibitors and cancer therapy. Radiother Oncol 2018;126(3):450-64.
- BIOVIA DS. Discovery Studio San Diego: Dassault Systèmes; 2017.
- BIOVIA DS. Pipeline pilot. San Diego: Dassault Systèmes; 2017.
- Huang S, Garbaccio RM, Fraley ME, Steen J, Kreatsoulas C, Hartman G, et al. Development of 6-substituted indolylquinolinones as potent Chek1 kinase inhibitors. Bioorg Med Chem Lett 2006;16(22):5907-12.
- Zhao L, Zhang Y, Dai C, Guzi T, Wiswell D, Seghezzi W, et al. Design, synthesis and SAR of thienopyridines as potent CHK1 inhibitors. Bioorg Med Chem Lett 2010;20(24):7216-21.
- Dwyer MP, Paruch K, Labroli M, Alvarez C, Keertikar KM, Poker C, et al. Discovery of pyrazolo[1,5-a]pyrimidine-based CHK1 inhibitors: A template-based approach-Part 1. Bioorg Med Chem Lett 2011;21(1):467-70.
- Spassov VZ, Yan L. A fast and accurate computational approach to protein ionization. Protein Sci 2008;17(11):1955-70.
- Gazzard L, Williams K, Chen H, Axford L, Blackwood E, Burton B, et al. Mitigation of Acetylcholine Esterase Activity in the 1,7-Diazacarbazole Series of Inhibitors of Checkpoint Kinase 1. J Med Chem 2015;58(12):5053-74.
- Gazzard L, Appleton B, Chapman K, Chen H, Clark K, Drobnick J, et al. Discovery of the 1,7-diazacarbazole class of inhibitors of checkpoint kinase 1. Bioorg Med Chem Lett 2014;24(24):5704-9.
- Fadrná E, Hlade?ková K, Ko?a J. Long-range electrostatic interactions in molecular dynamics: an endothelin-1 case study. J Biomol Struct Dyn 2005;23(2):151-62.
- Wu G, Robertson DH, Brooks III CL, Vieth M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J Comput Chem 2003;24(13):1549-62.
- Krammer A, Kirchhoff PD, Jiang X, Venkatachalam CM, Waldman M. LigScore: a novel scoring function for predicting binding affinities. J Mol Graph Model 2005;23(5):395-407.
- Muegge I. PMF scoring revisited. J Med Chem 2006;49(20):5895-902.
- Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 2015;10(5):449-61.
- Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol 2011;29:1039-45.
- Fraley ME, Steen JT, Brnardic EJ, Arrington KL, Spencer KL, Hanney BA, et al. 3-(Indol-2-yl)indazoles as Chek1 kinase inhibitors: Optimization of potency and selectivity via substitution at C6. Bioorg Med Chem Lett 2006;16(23):6049-53.
- Oza V, Ashwell S, Brassil P, Breed J, Ezhuthachan J, Deng C, et al. Synthesis and evaluation of triazolones as checkpoint kinase 1 inhibitors. Bioorg Med Chem Lett 2012;22(6):2330-7.
- Dudkin VY, Rickert K, Kreatsoulas C, Wang C, Arrington KL, Fraley ME, et al. Pyridyl aminothiazoles as potent inhibitors of Chk1 with slow dissociation rates. Bioorg Med Chem Lett 2012;22(7):2609-12.
- Garbaccio RM, Huang S, Tasber ES, Fraley ME, Yan Y, Munshi S, et al. Synthesis and evaluation of substituted benzoisoquinolinones as potent inhibitors of Chk1 kinase. Bioorg Med Chem Lett 2007;17(22):6280-5.
- Oza V, Ashwell S, Almeida L, Brassil P, Breed J, Deng C, et al. Discovery of Checkpoint Kinase inhibitor (S)-5-(3-Fluorophenyl)-N-(piperidin-3-yl)-3-ureidothiophene-2-carboxamide (AZD7762) by Structure-Based Design and Optimization of Thiophenecarboxamide Ureas. J Med Chem 2012;55(11):5130-42.
- Labroli M, Paruch K, Dwyer MP, Alvarez C, Keertikar K, Poker C, et al. Discovery of pyrazolo[1,5-a]pyrimidine-based CHK1 inhibitors: A template-based approach-Part 2. Bioorg Med Chem Lett 2011;21(1):471-4.
- Huang X, Cheng CC, Fischmann TO, Duca JS, Richards M, Tadikonda PK, et al. Structure-based design and optimization of 2-aminothiazole-4-carboxamide as a new class of CHK1 inhibitors. Bioorg Med Chem Lett 2013;23(9):2590-4.
- Massey AJ, Stokes S, Browne H, Foloppe N, Fiumana A, Scrace S, et al. Identification of novel, in vivo active Chk1 inhibitors utilizing structure guided drug design. Oncotarget 2015;6(34):35797-812.
- Osborne JD, Matthews TP, McHardy T, Proisy N, Cheung K-MJ, Lainchbury M, et al. Multiparameter Lead Optimization to Give an Oral Checkpoint Kinase 1 (CHK1) Inhibitor Clinical Candidate: (R)-5-((4-((Morpholin-2-ylmethyl)amino)-5-(trifluoromethyl)pyridin-2-yl)amino)pyrazine-2-carbonitrile (CCT245737). J Med Chem 2016;59(11):5221-37.
- Tong Y, Claiborne A, Stewart KD, Park C, Kovar P, Chen Z, et al. Discovery of 1,4-dihydroindeno[1,2-c]pyrazoles as a novel class of potent and selective checkpoint kinase 1 inhibitors. Biorg Med Chem 2007;15(7):2759-67.
- Yang B, Vasbinder MM, Hird AW, Su Q, Wang H, Yu Y, et al. Adventures in Scaffold Morphing: Discovery of Fused Ring Heterocyclic Checkpoint Kinase 1 (CHK1) Inhibitors. J Med Chem 2018;61(3):1061-73.
- Foloppe N, Fisher LM, Francis G, Howes R, Kierstan P, Potter A. Identification of a buried pocket for potent and selective inhibition of Chk1: Prediction and verification. Biorg Med Chem 2006;14(6):1792-804.
- Tao Z-F, Wang L, Stewart KD, Chen Z, Gu W, Bui M-H, et al. Structure-Based Design, Synthesis, and Biological Evaluation of Potent and Selective Macrocyclic Checkpoint Kinase 1 Inhibitors. J Med Chem 2007;50(7):1514-27.
- Reader JC, Matthews TP, Klair S, Cheung K-MJ, Scanlon J, Proisy N, et al. Structure-Guided Evolution of Potent and Selective CHK1 Inhibitors through Scaffold Morphing. J Med Chem 2011;54(24):8328-42.
- Matthews TP, Jones AM, Collins I. Structure-based design, discovery and development of checkpoint kinase inhibitors as potential anticancer therapies. Expert Opin Drug Discov 2013;8(6):621-40.
- Triballeau N, Acher F, Brabet I, Pin J-P, Bertrand H-O. Virtual Screening Workflow Development Guided by the “Receiver Operating Characteristic” Curve Approach. Application to High-Throughput Docking on Metabotropic Glutamate Receptor Subtype 4. J Med Chem 2005;48(7):2534-47.
- Li H, Leung K-S, Wong M-H, Ballester PJ. Correcting the impact of docking pose generation error on binding affinity prediction. BMC Bioinformatics 2016;17(11):308.
- Huang SY, Zou X. Efficient molecular docking of NMR structures: application to HIV-1 protease. Protein Sci 2007;16(1):43-51.