- *Corresponding Author:
- S. Gupta
Department of Nutrition,
Case Western Reserve University,
School of Medicine,
Cleveland,
Ohio 44106,
USA
E-mail: sanjay.gupta@case.edu
| Date of Received | 16 April 2020 |
| Date of Revision | 05 July 2021 |
| Date of Acceptance | 01 November 2021 |
| Indian J Pharm Sci 2021;83(6):1181-1195 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
Severe acute respiratory syndrome coronavirus 2 and associated coronavirus disease 2019 is a newly identified human coronavirus has imposed a serious threat to global health. The rapid transmission of severe acute respiratory syndrome coronavirus 2 and its ability to spread in humans have prompted the development of new approaches for its treatment. Severe acute respiratory syndrome coronavirus 2 requires RNA-dependent RNA polymerases for life cycle propagation and Spike (S)-protein for attachment to the host cell surface receptors. The virus enters the human body with the assistance of a key functional host receptor dipeptidyl peptidase-4 primed by transmembrane serine protease 2 which are putative targets for drug development. We performed screening of 267 compounds from Curcuma longa L. (Zingiberaceae family) against the viral S-protein and RNA-dependent RNA polymerases and host receptor proteins dipeptidyl peptidase-4 and transmembrane serine protease 2 using in silico molecular docking. Compounds C1, ((4Z,6E)-1,5-dihydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-4,6-dien- 3-one) and C6 ((4Z,6E)-1,5-dihydroxy-1-(4-hydroxy-3-methoxyphenyl)-7-(4-hydroxyphenyl)hepta-4,6- dien-3-one) exhibited tight binding to the S1 domain of the Spike protein than VE607 and with RNA- dependent RNA polymerase protein more effectively than ribavirin and remdesivir. These compounds also interacted with the human host proteins dipeptidyl peptidase-4 and transmembrane serine protease 2 with higher efficiency than standard inhibitors sitagliptin and camostat mesylate. The lead compounds showed favorable free binding energy for all the studied protein-ligand complexes in Molecular mechanics/ Generalized born model and solvent accessibility analysis. Besides, other Curcuma longa compounds C14 and C23 exhibited almost similar potential against these target proteins. The structure based optimization and molecular docking studies have provided information on some lead Curcuma longa compounds with probability for advancement in preclinical research.
Keywords
Severe acute respiratory syndrome coronavirus 2, Curcuma longa, transmembrane serine protease 2, dipeptidyl peptidase-4, spike glycoprotein, RNA dependent RNA polymerase
Coronavirus Disease 2019 (COVID-19) is an acute respiratory illness caused by a novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV -2)[1]. COVID-19 was first identified in December 2019, spreading from its likely origin in Wuhan, China and throughout the virus has affected millions of people worldwide. On March 11th, 2020, World Health Organization (WHO) announced the SARSCoV- 2 outbreak as pandemic with a state of public health emergency. As of May 7th, 2020, more than 3.77 million cases have been reported across 187 countries and territories, resulting in more than 265 000 deaths (https://www.worldometers.info/coronavirus/). According to current reports, SARS-CoV-2 transmission initiated from bats to humans, followed by human to human transmission occurring through small droplets produced while coughing, sneezing and talking or during close contact[2]. The rapidly increasing number of patients worldwide has prompted researchers to find treatment(s) and cure against this viral infection. Currently there is lack of therapy against SARSCoV- 2 infection, though several compounds have been investigated against some druggable targets for this disease.
The single-stranded SARS-CoV-2 virus enter cells via endocytosis through surface spike (S) proteins and bind to the Angiotensin-Converting Enzyme 2 (ACE- 2) and Dipeptidyl Peptidase-4 (DPP4/CD26) receptors. Binding of S-protein to ACE-2 triggers a conformational change in the S-protein facilitating proteolytic digestion by Transmembrane Serine Protease 2 (TMPRSS2) and allows fusion of virus to the cell membrane[3]. Upon entering the host cell, the viral particle is decoded and assembled for translation utilizing the Open Reading Frames 1a (ORF1a) and Open Reading Frames 1ab (ORF1ab) generating Polyproteins (pp1a and pp1ab). These polyproteins undergo cleavage to form structural proteins for the Ribonucleic acid (RNA) replicasetranscriptase complex responsible for the replication and transcription of viral RNA[4]. The disruption of the replication processes could lead to identification of potential molecular target(s) to develop effective treatment strategies.
It has been known that many viruses lack preventive vaccines and/or potential antiviral therapy because of the high rate of genomic mutation in the virus allowing them to rapidly evolve and adapt to the host environment[5]. Under these circumstances traditional herbal medicines and phytochemicals are excellent source for antiviral drug discovery. Some studies demonstrated that natural phytochemicals have different kinds of activities against microorganism and germs including viruses[6-8]. Natural compounds such as scutellarein, silvestrol, tryptanthrin, amentoflavone, quercetin, myricetin and lectins have shown promise in the suppression of viral attachment and inhibitors of viral enzymes such as proteases, helicases and polymerases[9,10]. Griffithsin, a liptin derived from red algae inhibits the binding of SARSCoV- 2 spike protein with the human ACE-2 receptor (viral attachment) by virtue of its Receptor-Binding Domain (RBD) (glycosylation site in S1 subunit of spike protein) binding potential[11]. Previously, Keyaerts et al. showed the SARS-CoV spike protein mannose binding potential of Hippeastrum Hybrid Agglutinin (HAA) lectin in vitro[12]. In another study, Cheng et al. reported that saikosaponin B2 natural compound has anticoronaviral activity at micromolar concentrations.
The study suggested that saikosaponin B2 possess viral attachment and penetration inhibition potential[13]. Curcuma longa L. (C. longa) (Zingiberaceae family) is a well-known medicinal plant in Ayurveda and other traditional medicinal systems[14]. It has been used as a dietary spice and herbal supplement. Phytochemicals present in C. longa possess various pharmacological properties including antiviral activity against dengue and hepatitis C virus[15,16] and have demonstrated suppression of infection against Zika and Chikungunya virus through inhibition of binding at the cell surface[17]. Curcumin and related compounds present in C. longa inhibit Human Immunodeficiency Virus (HIV) replication by targeting HIV-1 integrase and HIV-2 proteases[13]. Curcumin, demethoxycurcumin and bisdemethoxycurcumin inhibit attachment of influenza virus by targeting neuraminidase protein[18]. Recently our research group reported that C. longa phytochemicals have potential to inhibit SARS-CoV-2 viral virulence by inhibiting Main Protease (Mpro) enzyme[19].
In the present study, we screened a small library of compounds from C. longa against SARS-CoV-2 proteins viz. spike glycoprotein (S-protein) and RNA dependent RNA polymerase (RdRp) and human host proteins including CD26 and TMPRSS2 as potential targets and drug designing candidates against SARS- CoV-2
Materials and Methods
C. longa compound retrieval and preparation:
A total of 267 compounds present in C. longa plant were obtained from different literature and search engine platforms such as PubMed, Google Scholar, Web of Science, Science Direct, Scopus, Semantic Scholar, Medline and PubMed Central[20]. The structures of compounds present in C. longa were prepared by using Marvin Sketch software[21]. The Two Dimensional (2D) or Three Dimensional (3D) structure of standard compounds against targeted proteins was retrieved from the National Center for Biotechnology Information (NCBI) PubChem in Spatial Data File (.sdf) format[22]. Open Babel molecule format converter was used to perform conversion of 2D to 3D conformation and their conversion from .sdf to Molecular (.mol) data file[23]. Ligand energy was minimized by applying Merck Molecular Force Field (MMFF94) and conjugate gradients optimization algorithm using PyRx-Python prescription 0.8 for 200 steps[24].
Receptor retrieval:
3D structure of SARS-CoV-2 S-protein (Protein Data Bank (PDB) ID: 6VSB) and human CD26 (PDB ID: 4PNZ) receptor were obtained from PDB (https://www. rcsb.org/)[25]. The resolutions of the retrieved structures were between 1.9 Å to 3.46 Å.
Homology modeling of RdRp and TMPRSS2 protein:
The crystal structure of SARS-CoV-2 RdRp and human TMPRSS2 protein has not been elucidated. Thus, the 3D structure of these two proteins was modeled using the Swiss-model structural bioinformatics server[26]. Amino Acid (AA) sequences of SARS-CoV-2 RdRp (NCBI reference sequence: YP_009725307.1) and TMPRSS2 (NCBI reference sequence: NP_001128571.1) were retrieved from the NCBI database. We used SARS coronavirus Non-Structural Protein 12 (NSP12) (PDB ID-6NUR) and serine protease hepsin (PDB ID-5CE1) as respective templates for modeling of SARS-CoV-2 RdRp and TMPRSS2. For the alignment of TMPRSS2 and RdRp target with their respective template sequences the T-Coffee server was used[27]. Representation of the alignment was made using ESPript 3.0 server[28]. Modelled structures were refined using ModRefiner sever[29].
Receptor preparation:
3D structure of SARS-CoV-2 S-protein and human CD26 protein was loaded onto the University of California, San Francisco (UCSF) Chimera for molecular docking preparation[30]. Protein models were cleaned and optimized by removing ligands and other heteroatoms including water. After this step, the energy minimization of protein structures was performed by steepest descent method having 100 steps (step size 0.02 Å) and a conjugate gradient method with 10 steps (step size 0.02 Å) using UCSF Chimera.
Active site prediction of RdRp and TMPRSS2 modelled protein:
It is anticipated that an effective drug ligand to dock either within the protein pocket or the functional active region. The possible binding sites were searched for RdRp and TMPRSS2 modeled proteins using Computer Atlas of Surface Topography of Proteins (CASTp) 22 tool. The CASTp22 tool predicts potential pockets of target protein and confirms whether the highest frequency binding sites of the heat map were located within the protein pocket[31].
Molecular docking studies:
Auto Dock Tools 1.5.6 (ADT) was used to dock the test ligands on targeted protein[32]. Gasteiger partial charges assigned to the ligands and docking calculations were performed. Polar hydrogen atoms, Kollman charges and solvation parameters were applied using appropriate Auto Dock tool. Lamarckian Genetic Algorithm (LGA) was used to explore the active binding region in this study. The grid box included the entire binding site of the protein providing enough space for the ligands translational and rotational walk. For each of the 30 independent runs, a maximum number of 27 000 Genetic Algorithm (GA) operations were generated on a single population of 150 individuals. Operator weights for the rate of crossover, gene mutation and elitism were set as 0.80, 0.02 and 1 respectively. LigPlot+ (v.1.4.5) and UCSF chimera (v.1.10.2) online tools were used for protein-ligand interaction visualization[33].
Molecular Mechanics/Generalized Born Model and Solvent Accessibility (MM-GBSA) analysis:
Prime MM-GBSA analysis was used to evaluate the receptor/protein and receptor/protein-ligand binding energies, which includes the VSGB solvent model, Optimized Potentials for Liquid Simulations (OPLS)- 2005 force field and rotamer search algorithms. The Prime MM-GBSA simulation was performed by using the Glide pose viewer file to compute the total free energy of binding. The MM-GBSA calculations were attained to evaluate the relative binding affinity of test molecules and their respective complexes with lead ligands (reported in kcal/mol). As the MM- GBSA binding energies are approximate free energies of binding, a more negative value indicates stronger binding.
Results and Discussion
In the present study, a total of 267 C. longa compounds were searched from the literature and docked against two SARS-CoV-2 proteins including S-protein and RdRp and two human host proteins CD26 and TMPRSS2. Binding score of lead compounds against SARS-CoV-2 S-protein and RdRp modelled, CD26 and TMPRSS2 modelled proteins are shown in fig. 1A-fig. 1D with a cut-off score of £-7, £-4, £-6 and £-5 kcal/mole. The structure of lead compounds with their name and targets are represented in Table 1[34-47]. CASTp server was utilized to predict the binding pockets in RdRp and TMPRSS2 modeled protein. The predicted solvent-accessible surface area and volume for the RdRp protein was 2796.437 Å2 and 5657.063 Å3 respectively. Similarly, the predicted solvent-accessible surface area and volume for the TMPRSS2 protein was 338.267 Å2 and 264.585 Å3 respectively (fig. 2A and fig. 2B).
| S. No | Compound name | Structure | Reference | Targeted protein |
|---|---|---|---|---|
| 1 | (4Z,6E)?1,5?dihydroxy?1,7?bis(4?hydroxy?3?methoxyphenyl)hepta?4,6?dien?3?one |
|
[28] | Spike#, CD26, TMPRSS2*, RdRp* |
| 2 | (1E)?1,7?bis(4?hydroxy?3?methoxyphenyl)hept?1?ene?3,5?dione |
|
[29,30] | Spike, CD26, TMPRSS2 |
| 3 | Tetrahydroxycurcumin |
|
[31] | Spike, CD26, TMPRSS2, RdRp |
| 4 | (1E)?7?(4?hydroxy?3?methoxyphenyl)?1?(4?hydroxyphenyl)hept?1?ene?3,5?dione |
|
[29,30] | CD26 |
| 5 | (1E)?1,7?bis(4?hydroxyphenyl)hept?1?ene?3,5?dione |
|
[29,30] | CD26 |
| 6 | (4Z,6E)?1,5?dihydroxy?1?(4?hydroxy?3?methoxyphenyl)?7?(4?hydroxyphenyl)hepta?4,6?dien?3?one |
|
[28] | Spike, CD26, TMPRSS2, RdRp |
| 7 | 2?methyl?5?(6?methylhept?5?en?2?yl)cyclohex?2?ene?1,4?diol |
|
[32] | CD26 |
| 8 | (4Z,6E)?1,5?dihydroxy?7?(4?hydroxy?3?methoxyphenyl)?1?(4?hydroxyphenyl)hepta?4,6?dien?3?one |
|
[28] | Spike, CD26 |
| 9 | (6E)?3?hydroxy?1,7?bis(4?hydroxyphenyl)hept?6?ene?1,5?dione |
|
[28] | Spike, CD26, TMPRSS2, RdRp |
| 10 | Cyclocurcumin |
|
[33] | CD26 |
| 11 | (4Z,6E)?1,5?dihydroxy?1,7?bis(4?hydroxyphenyl)hepta?4,6?dien?3?one |
|
[28] | Spike, CD26, TMPRSS2, RdRp |
| 12 | Santalol A |
|
[34] | CD26 |
| 13 | Bisacurone |
|
[28] | CD26, RdRp |
| 14 | 5?hydroxy?6?(3?hydroxy?4?methylphenyl)?2?methylhept?2?en?4?one |
|
[35] | CD26, RdRp |
| 15 | Bisacurone B |
|
[36] | CD26, RdRp |
| 16 | 6?(5?hydroxy?2?methoxy?4?methylcyclohex?3?en?1?yl)?2?methylhept?2?en?4?one |
|
[37] | CD26 |
| 17 | 2?hydroxy?4?(6?methyl?4?oxohept?5?en?2?yl)benzaldehyde |
|
[37] | CD26 |
| 18 | (1E,6E)?1?(3,4?dihydroxyphenyl)?7?(4?hydroxyphenyl)hepta?1,6?diene?3,5?dione |
|
[37] | TMPRSS2, RdRp |
| 19 | (1E,4E)?1,5?bis(4?hydroxy?3?methoxyphenyl)penta?1,4?dien?3?one |
|
[32] | TMPRSS2 |
| 20 | (1E,4E,6E)?1,7?bis(4?hydroxyphenyl)hepta?1,4,6?trien?3?one |
|
[38] | TMPRSS2 |
| 21 | 1?(5?hydroxy?3,6?dimethyl?2,3,3a,4,5,7a?hexahydro?1?benzofuran?2?yl)?3?methylbut?2?en?1?one |
|
[32] | TMPRSS2 |
| 22 | (1E)?1?(4?hydroxy?3?methoxyphenyl)?7?(4?hydroxyphenyl)hept?1?ene?3,5?dione |
|
[29,30] | TMPRSS2 |
| 23 | Calebin A |
|
[30] | TMPRSS2, RdRp |
| 24 | Curcumin |
|
[39] | Spike |
| 25 | 1,6-Heptadiene-3,5-dione, 1,7-bis(4-hydroxy-3-methoxyphenyl) |
|
[40] | Spike |
| 26 | Curculonone D |
|
[32] | RdRp |
| 27 | Vanillin |
|
[41] | RdRp |
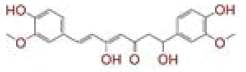
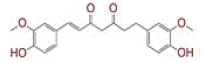
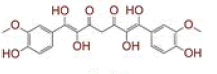
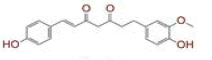
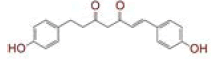
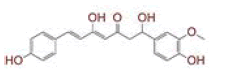
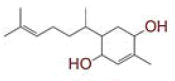
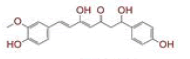
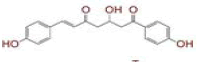
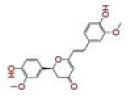
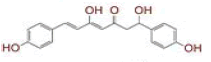
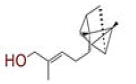
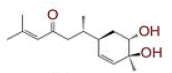
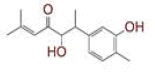
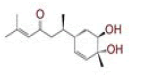
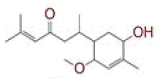
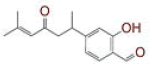
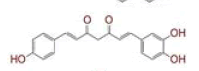
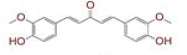
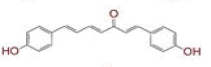
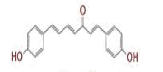
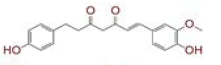
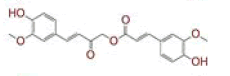
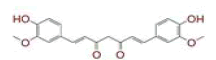
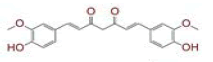
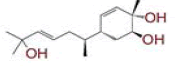
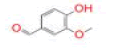
Table 1: List of Lead C. Longa Phytochemicals Against Sars-Cov-2 and Human Host Proteins Related to Viral Infection

Fig. 1: Docking score of C. longa compounds against SARS-CoV-2 virus and human host proteins, (A) Docking score of lead compounds (≤-6 kcal/mol) against human CD26 protein; (B) Docking score of lead compounds (≤-5 kcal/mole) against modeled human TMPRSS2 protein; (C) Docking score of lead compounds (≤-7 kcal/mole) against SARS-CoV-2 spike glycoprotein; (D) Docking score of lead compounds (≤-4 kcal/mole) against SARS-CoV-2 RdRp; (E) Structure of lead C. longa compound [(4Z,6E)-1,5- dihydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-4,6-dien-3-one] against human CD26 and SARS-CoV-2 spike glycoprotein;(F) Structure of lead C. longa compound [(4Z,6E)-1,5-dihydroxy-1-(4-hydroxy-3-methoxyphenyl)-7-(4-hydroxyphenyl)hepta-4,6- dien-3-one] against human TMPRSS2 and RdRp modeled protein; (a) Sitagliptin; (b) Vildagliptin; (c) Linagliptin; (d) Saxagliptin;(e) Alogliptin; (f) Teneligliptin; (a’) Ribavirin; (b’) Remdesivir; (c’) Galidesivir; (d’) Tenofovir; (e’) Sofosbuvir; CM-Camostat mesylate

Fig. 2: The surface of the ligand binding pocket calculation of (A) RdRp and (B) TMPRSS2 modeled protein using CASTp 3.0. server. The binding pocket is shown in red color
The in silico models SARS-CoV-2 RdRp and human TMPRSS2 proteins were generated by homology modeling employing the Swiss Model web server and CASTp22 prediction tool[26,31]. The homology model of the TMPRSS2 was adopted from our recently published article[47]. Alignment analysis of the RdRp and TMPRSS2 with their respective template sequences utilized for the homology modelling is depicted in fig. 3. The generated modelled proteins were respectable at different modelling parameters such as MolProbity score, Clash score, local similarity to the target, normalized QMEAN score, Ramachandran favored, Ramachandran outliners, Bad angles and Bad bonds (fig. 4 and Table 2)[48]. AA residue of targeted proteins (PDB 4PNZ, PDB 6VSB; RdRp modeled and TMPRSS2 modeled), type of interaction, binding energy of protein-ligand complex and binding affinity of some lead compounds against respective protein has been summarized in Table 3. MM-GBSA analysis of the lead C. longa phytochemicals and standard inhibitors with their respective protein/receptor complexes and unbound proteins was performed and the results are shown in Table 4. Results showed favorable free energy of protein/receptor-ligand complexes in comparison to unbound proteins in MM-GBSA analysis. Various standard inhibitors from published studies were engaged to compare the target protein binding potential of C. longa compounds[49-53] (fig. 1A-fig. 1D. Binding pattern of the compounds against viral and human host proteins are shown in fig. 5 and fig. 6. Docking score of standard compound against SARS-CoV-2 S-protein,RdRp, CD26 and TMPRSS2 proteins are shown in fig. 1A-fig. 1D. Result showed that compound C1 (-7.88 kcal/mole) and C6 (-6.62 kcal/mole) showed minimum binding score against CD26 and TMPRSS2 proteins. Compound C1 ((4Z,6E)-1,5-dihydroxy-1,7- bis(4-hydroxy-3-methoxyphenyl)hepta-4,6-dien-3- one) showed Hydrogen Bonding (HB) and hydrophobic interaction pattern with the CD26 protein (Table 3). Compound C6 ((4Z,6E)-1,5-dihydroxy-1-(4-hydroxy- 3-methoxyphenyl)-7-(4-hydroxyphenyl)hepta-4,6-dien-3-one) showed disulfide/HB, hydrophobic and pi-pi interaction pattern with the modeled TMPRSS2 protein (Table 3).
| Parameters | TMPRSS2 protein | RNA polymerase protein |
|---|---|---|
| MolProbity score | 1.78 | 1.03 |
| Clash score | 5.45 | 0.85 |
| Ramachandran favored | 92.15 % | 96.05 % |
| Ramachandran outliers | 0.87 % | 0.56 % |
| Bad bonds | 0/2785 | 0/7370 |
| Bad angles | 31/3791 | 47/9999 |
Table 2: Protein Modelling Parameters and Their Values
| Protein | CN | AA residue involve in HB | AA residue involvein HI/DSI/Pi-Pi interactions |
|---|---|---|---|
| CD26 | C1 | Asp739, Asp545, Asn710, Lys554 | Glu205, Asn709, Trp201, Arg125, Trp124, Lys122, His740, Gly741, Ala743, Tyr547, Val546, Trp627, Gly628, Tyr752, Trp629, Ser630 |
| C2 | NF | Asp739, ASH709, Asp545, Lys554, Asn710, Lys122, Trp124, Arg125, His740, Gly741, Glu205, Trp201, Tyr752, Val546, Tyr547, Trp627, Gly628, Trp629, Ser630, Gly632 | |
| C3 | Asn710, ASH709, Asp739, Lys554 | Glu205, Trp201, His740, Gly741, Lys122, Trp124, Arg125, Trp629, Gln553, Ser552, Tyr547 | |
| TMPRSS2 | C6 | NF | Leu188, Gly190, Phe193, Val283, Ala280, Ile279, Arg277, Ile489, Thr324, Pro325, Cyx278*, Cyx281*, Tyr227, Phe231 |
| C2 | Tyr227, Arg277 | Phe231, Phe193, Gly190, Tyr189, Leu188, Ile489, Val283, Ala280, Ile279, Pro325, Thr324, Pro400, Lys399, Phe394, Cyx281*, Cyx278* | |
| C11 | Leu188 | Phe193, Gly190, Thr227, Arg277, Ile279, Ala280, Pro335, Thr324, Cyx278*, Phe231 | |
| C3 | Tyr227 | Ile489, Arg277, Ala280, Ile279, Val283, Leu188, Tyr189, Gly190, Phe193, Phe394, Pro400, Pro325, Thr324, Phe231, Cyx278*, Cyx281* | |
| C18 | Tyr227, Arg277, Ala280 | Leu188, Gly190, Phe193, Pro325, Thr324, Pro400, Ala280, Ile279, Cyx281*, Cyx278*, Phe394 | |
| Spike | C1 | Gln493, Tyr351, Val350, Val401 | Lys417, Ile418, Asn422, Leu492, Gln493, Ser494, Tyr351, Tyr495, Val350, Ser349, Phe347, Ile402, Asp442, Arg509, Phe497 |
| C8 | Val401, Val350, Tyr351, Pro491, Gln493, | Tyr495, Ser494, Gln493, Leu492, Lys417, Ile418, Tyr421, Asn422, Ser349, Phe347, Asp442, Phe497 | |
| C2 | Phe347, Val350, Tyr495 | Asp442, Ala348, Ser349, Tyr351, Lys417, Ile418, Tyr421, Asn422, Pro491, Leu492, Gln493, Ser494, Phe497, Val401,Arg509, Tyr495 | |
| C9 | Tyr351, Gln493, Tyr495, Asp442 | Pro491, Leu492, Ser494, Phe497, Arg509, Val401, Phe347, Ala348, Ser349, Val350, Asn422, Lys417, Ile418 | |
| C6 | Gln493, Pro491, Tyr351, Val350, Val401 | Ile402, Ser349, Asn422, Leu492, Ser494, Tyr495, Asp442, Ile418, Lys417, Phe497 | |
| RNA polymerase | C6 | Asp623, Asp618, Asp761 | Arg624, Cys622, Lys621, Pro620, Tyr619, Trp617, Gly616, Arg553, Lys551, Lys798, Cys799, Trp800, Asp760, Ala762, Glu811, Phe812 |
| C1 | Asp623, Asp760, Asp761, Ser814, Lys798 | Asn691, Thr680, Cys622, Arg553, Arg555, Tyr619, Asp618, Trp617, Gly616, Cys799, Trp800, Glu811, Phe812, Cys813, Ser759 | |
| C14 | Ser814, Glu811, Asp761, Trp800 | Cys813, Phe812, Cys799, Lys798, Gly616, Trp617, Asp618, Ala762, Asp760, Ser759, Leu758 | |
| C23 | Thr556, Lys798 | Val763, Ala762, Asp761, Asp760, Arg624, Asp623, Arg553, Asp452, Arg555, Val557, Lys545, Gly616, Trp617, Asp618, Phe812, Glu811, Trp800, Cys799 |
Note: HB-Hydrogen bond; HI-Hydrophobic interaction; *-indicates residues with Pi-Pi interaction; DSI-Digital sequence information
Table 3: Sars-Cov-2 and Human Host Protein AA Residues Involved in The Interaction with C. longa Compounds
| Receptor name | Receptor/Protein energy | Receptor/Protein-standard ligand complex energy | Receptor/Protein-lead ligand complex energy |
|---|---|---|---|
| CD26 | -63281.79373 | -63286.47008 | -63381.1174 |
| Spike | -128584.6622 | -128598.4453 | -128681.7845 |
| RNA pol | -33846.13982 | -33825.36073 | -33957.28006 |
| TMPRSS2 | -13641.03537 | -13788.34184 | -13772.66361 |
Note: All the numerical values are shown in the table are given in kcal/mole. Sitagliptin, VE607, Ribavirin and CM were used as standard ligand for CD26, Spike, RdRp and TMPRSS2 proteins respectively. C. longa compound [(4Z,6E)?1,5?dihydroxy?1,7?bis(4?hydroxy?3?methoxyphenyl)hepta?4,6?dien?3?one] was the lead ligand for CD26 and SARS-CoV-2 spike glycoprotein. C. longa compound [(4Z,6E)?1,5?dihydroxy?1?(4?hydroxy?3?methoxyphenyl)?7?(4?hydroxyphenyl)hepta?4,6?dien?3?one] was the lead ligand for TMPRSS2 and RdRp
Table 4: Mm-Gbsa Analysis of the Sars-Cov-2 Targeted Protein and Protein-Standard Inhibitor/Lead Molecule Complex

Fig. 3: Alignment of the target protein with the template protein sequence, (A) Alignment of RdRp with the template and (B) Alignment of TMPRSS2 with the template

Fig. 4: Modelling parameters of SARS-CoV-2 RdRp modeled protein, (A) Ramachandran plot; (B) Local quality estimate and (C) Z-score of the modeled protein

Fig. 5: Interaction of C. longa compounds with CD26 and TMPRSS2 human host proteins, (A) Surface structure and type of interaction involved in the compound C1 ((4Z,6E)-1,5-dihydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-4,6-dien-3-one) and CD26 protein interaction. Protein and ligand are shown in red and yellow color; (B) Surface structure and type of interaction involved with compound C6 ((4Z,6E)-1,5-dihydroxy-1-(4-hydroxy-3-methoxyphenyl)-7-(4-hydroxyphenyl)hepta-4,6-dien-3-one) and TMPRSS2 modeled protein. Protein and ligand are shown in cyan and purple color

Fig. 6: Interaction of C. longa compounds with SARS-CoV-2 and RdRp proteins, (A) Surface structure and type of interaction involved with compound C1 ((4Z,6E)-1,5-dihydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-4,6-dien-3-one) and SARS-COV-2 spike glycoprotein. Protein and ligand are shown in yellow and red color; (B) Surface structure and type of interaction involved with the compound C6 ((4Z,6E)-1,5-dihydroxy-1-(4-hydroxy-3-methoxyphenyl)-7-(4-hydroxyphenyl)hepta-4,6-dien-3-one) and RdRp modeled protein. Protein and ligand are shown in green and red color
Camostat mesylate (CM) is a known, in vitro validated human TMPRSS2 protein inhibitor[53,54]. We predicted the active site (ligand binding site) of the TMPRSS2 modeled protein. Docking of CM and C1 (4Z,6E)- 1,5-dihydroxy-1-(4-hydroxy-3-methoxyphenyl)-7-(4- hydroxyphenyl)hepta-4,6-dien-3-one was performed against the site to predict the comparative binding of both compounds. The result showed that C. longa compound binds more tightly (-6.62 kcal/mole) to the TMPRSS2 predicted active site in comparison to known CM inhibitor (-5.14 kcal/mole) (fig. 7A and fig. 7B). Moreover, the lead ligand bound structures of the CD26 and modeled TMPRSS2 protein showed slight changes in Root-Mean-Square Deviation (RMSD) values (0.304Å and 0.371Å) in comparison to unbound structure (fig. 8A-fig. 8E).

Fig. 7: Binding of lead and standard compounds at predicted binding site at TMPRSS2 protein compared with the unbound protein inhibitors at allosteric and predicted active site of the protein, (A) Binding of (4Z,6E)-1,5-dihydroxy-1-(4-hydroxy-3-methoxyphenyl)- 7-(4-hydroxyphenyl)hepta-4,6-dien-3-one phytochemical (red color) at TMPRSS2 predicted active site with binding score;
(B) Binding of in vitro validated TMPRSS2 inhibitor CM (red color) at predicted active site and its binding score. Ligand bound (yellow color) and unbound (cyan color) protein structures were superimposed to show the change in protein conformation. Change in ligand bound and unbound protein conformation is provided in the form of RMSD value and binding score of ligand at the predicted active site is given in kcal/mole

Fig. 8: C. longa compounds bound and un-bound 3D superimposed structure of SARS-CoV-2 and human host protein. Yellow color indicates unbound protein structure and ligand bound protein structure is shown in cyan color, (A) Superimposed ligand ((4Z,6E)-1,5-dihydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-4,6-dien-3-one) bound and unbound CD26 protein structures;
(B) Superimposed ligand ((4Z,6E)-1,5-dihydroxy-1-(4-hydroxy-3-methoxyphenyl)-7-(4-hydroxyphenyl)hepta-4,6-dien-3-one) bound and unbound TMPRSS2 modeled protein structures; (C) Superimposed ligand ((4Z,6E)-1,5-dihydroxy-1,7-bis(4-hydroxy- 3-methoxyphenyl)hepta-4,6-dien-3-one) bound and unbound SARS-CoV-2 spike glycoprotein structures; (D) Superimposed ligand ((4Z,6E)-1,5-dihydroxy-1-(4-hydroxy-3-methoxyphenyl)-7-(4-hydroxyphenyl)hepta-4,6-dien-3-one) bound and unbound RdRp modeled protein structures; (E) Difference in RMSD values of ligand bound and un-bound proteins
Compound C1 ((4Z,6E)-1,5-dihydroxy-1,7-bis (4-hydroxy-3-methoxyphenyl)hepta-4,6-dien-3-one) exhibited potential binding (-8.51 kcal/mole) at SARS-CoV-2 S1 domain and ACE-2 protein binding interface. Result showed HB and hydrophobic interactions between compound C1 and S1 domain (Table 2 and fig. 6A). Besides S-protein, we also targeted SARS- CoV-2 RdRp protein. Results showed that C. longa compounds (C1, C6, C14 and C23) have potential to bind SARS-CoV-2 RdRp approximately with similar potential ranging from -5.57 to -5.10 kcal/mole. Compound C1 interacts with RdRp protein through HB and hydrophobic interaction with a minimum binding score among all screened compounds (Table 3 and fig. 6B).
In the next experiment, the comparative docking pose of respective standard inhibitors against target proteins were studied (fig. 9A-fig. 9E). AA residues and type of interactions involved with the standard inhibitor- target protein binding is summarized in Table 3. Result showed that docking pose of standard inhibitor and lead compounds against CD26 protein were different (fig. 9A and Table 3). Compound C6 and CM interacted with the same AA residues viz. Arg277, Tyr227, Ile279, Thr324, Pro325, Phe231, Ala280, Phe193, Cys278, Gly190 and Leu188 of TMPRSS2 protein; although the type of interaction was different (fig. 9B and Table 3). Additional interactions noted with compound C6 were with Leu188, Gly190, Phe193, Val283, Ala280, Pro325, Cys281 and Phe231 AA residues. Compound C1 and VE607 showed similar pattern of binding with AA resides viz. Gln493, Tyr495, Ser494, Asn422, Leu492, Tyr351, Val350, Asp442 and Phe497 against SARS- CoV-2 S-protein (Table 2). Moreover, compound C1 showed additional binding with Lys417, Ile418, Tyr351, Val350, Ser349, Phe347, Ile402, Arg509 and Val401 residues (Table 3). Ribavirin, remdesivir (prodrug) and nucleoside triphosphate (active metabolite of remdesivir) were engaged as SARS RdRp standard inhibitors for docking pose comparison with C. longa compounds (Table 3). Similar to ribavirin, compound C6 showed interactions with Ala762, Asp761, Glu811, Lys798, Trp617, Asp760, Trp800 and Cys799 residue of SARS-CoV-2 RdRp enzyme (Table 3). Compound C6 showed binding with some additional AA residues including Asp623, Asp618, Arg624, Cys622, Lys621, Pro620, Tyr619, Gly616, Arg553, Lys551 and Phe812, respectively (Table 3). It should be noted that lead compound C6 and recently proposed SARS-CoV-2 RdRp inhibitor remdesivir prodrug and its active metabolite nucleoside triphosphate exhibited similar binding pattern on protein active site with binding energy -5.81 and -6.85 kJ/mole (fig. 9E). Compound C6 and remdesivir active drug (nucleoside triphosphate) interacted with almost similar AA residues including Asp618, Asp761, Lys621, Pro620, Trp617, Gly616,Lys551, Lys798, Trp800 and Glu811 (Table 3).

Fig. 9: Superimposed C. longa compounds and standard inhibitors at active site of SARS-CoV-2 and human host target proteins,
(A) Binding of compound C1 (green) and sitagliptin (red) at human host CD26 active site; (B) Binding of compound C6 (yellow) and CM (warm pink) at human host TMPRSS2 active site; (C) Binding of compound C1 (green) and VE607 (blue) at SARS-CoV-2 spike glycoprotein active site; (D) Binding of compound C6 (yellow) and ribavirin (cyan) at SARS-CoV-2 RNA-polymerase active site; (E) Binding of compound C6 (yellow), remdesivir prodrug (blue) and remdesvir active drug (nucleoside triphosphate) (red) at SARS-CoV-2 RNA-polymerase active site
Virus entry in the human cells is initiated through the interaction of viral protein with the human receptor(s) followed by conformational change in the viral protein which induces the internalization process[55]. Thus, the agents which can prevent the viral entry and their attachment to the host cells are important candidates for anti-viral drug discovery. Several natural products show their anti-viral potential by inhibiting the viral attachment. Several natural compounds show their anti- viral potential by inhibiting the viral attachment to the host cell. For example, natural tannin compounds viz. chebulagic acid and punicalagin inhibit the attachment and fusion of different viruses with the host receptor by deactivating the free viral particles[56]. It has been reported that some natural compounds including epigallocatechin-3-gallate a major constituent of green tea, has potential to inhibit the viral attachment, its replication, assembly or release of their progeny virions. The antiviral mechanism inhibits cell-to-cell spread of the virus and lowers the infectivity[57]. In the present study, we identified the binding potential of C. longa phytochemicals with the SARS-CoV-2 and human proteins involve in the viral-host protein interaction, internalization and replication process.
CD26, also known as DPP4, is a 110 kDa human cell- surface glycoprotein that exerts varying functions in cell type and physiological conditions in context-based manner. Lu et al. (2013) demonstrated that Middle East Respiratory Syndrome Coronavirus (MERS-CoV) spike protein bound with CD26 mediates attachment and fusion to host cells thereby initiating infection[54]. Human CD26 is an important immuno-regulator used by viruses for immune hijacking and virulence. TMPRSS2 is a serine protease that proteolytically cleaves and activates viral spike protein and facilitates virus-cell membrane fusion. It has been reported that TMPRSS2 plays a critical role in the proteolytic activation of SARS-CoV and MERS-CoV[58]. Therefore, targeting human CD26 and TMPRSS2 proteins is an efficient step to inhibit the cellular entry and virulence of SARS- CoV-2.
Our study corroborates with previous publications demonstrating binding of CD26 inhibitors at Tyr125, Glu205, Lys122, Trp124 and Ap739 amino residue of CD26[59,60]. In the present study, C. longa compounds binds to the predicted ligand binding (fig. 2B) of TMPRSS2 protein. Our study also corroborates with the recently published data on remdesivir which binds at the similar binding site of TMPRSS2[4]. The predicted active site of the modeled TMPRSS2 protein was relatively involved in the SARS-CoV-2 S-protein and TMPRSS2 protein interaction interface. Comparison of ligand and CM bound TMPRSS2 modeled protein showed that the binding of lead compound at the predicted site of the protein binds more tightly than the binding of CM (fig. 7A and fig. 7B). Thus, the comparative results indicate the TMPRSS2 binding potential of C. longa compounds at the predicted site. Minor change in RMSD values of ligand bound and unbound CD26 and TMPRSS2 structures indicate stable docking pose of C. longa compounds at respective binding sites of the protein.
SARS-CoV-2 spike glycoprotein is composed of S1 and S2 subunits[61]. The S1 and S2 domains are involved in host cell receptor binding and membrane fusion. The S1 subunit comprise of a signal peptide, N-terminal domain and RBD. The S1 domain interacts with human ACE-2 receptor and forms a fusion core that passages viral and human cellular membrane to initiate fusion and infection. Beside ACE-2 receptor, the S1 domain (RBD) of the spike protein interacts with several other host proteins such as CD26 and TMPRSS2. S2 subunit of SARS-CoV-2 S-protein consists of conserved fusion peptide mediating membrane fusion process[62]. Recent studies indicated that similar to SARS-CoV and MERS-CoV, the S1 domain of SARS-CoV-2 possesses conserved sequences which interact with human proteins underlying cell adhesion and virulence[63].
Tai et al. characterized the receptor binding domain of SARS-CoV-2 and reported the variable amino residues (331aa-524aa) among SARS-CoV-2 and SARS-CoV[64]. Wrapp et al. reported that S1 subunit of SARS-CoV-2 has 10-20 fold higher binding affinity for ACE-2 receptor than SARS-CoV which may contribute to its higher infectivity and transmission potential in comparison to SARS-CoV[65]. Tight binding of compound C1 at SARS-CoV-2 S1 domain and ACE-2 protein binding interface indicates the disruption of protein-protein interaction between these two molecules (fig. 5A). Our study corroborates with the recent findings showing the binding of hesperidin, a flavanone glycoside found in citrus fruits, at the SARS-CoV-2 S1 domain and ACE- 2 protein binding interface[60]. It should be noted that the compound C1 binds with some key AA residues (Gln493, Ile402, Lys417, Gln493 and Ser494) which are unique to the SARS-CoV-2 S1 domain. This result indicates the selective SARS-CoV-2 S1 domain targeting potential with C. longa compounds might disrupt the viral adherence on human host cells. Since polymerases play a central role in viral genome synthesis, replication and transcription, thus, targeting SARS-CoV-2 RdRp by C1 could emerge as a potential antiviral drug. Tight binding of other C. longa compounds C1, C6, C14 and C23 at SARS-CoV-2 RdRp active site indicates that the compounds might have inhibitory potential against viral replication.
In our study, similar binding pattern of compound C6 was noted as compared to standard inhibitor CM interaction with TMPRSS2 protein (fig. 7A, fig. 7B and fig. 8B). Binding with an additional AA residue might be the reason behind lower binding energy of compound C6 against TMPRSS2 protein than standard inhibitor (fig. 1). It has been reported that VE607 compound binds with SARS-CoV S-protein pseudotype HIV- 1 virus and thereby inhibit the viral entry in ACE-2 expressing 2393T cells[66]. Binding of compound C1 with additional residues of spike protein S1 domain might be responsible for its lower binding energy than VE607 compound (Table 3). Similar to SARS-CoV-2 RdRp inhibitors, compound C6 interacted with similar AAs. Altogether, C. longa compound show additional binding with the protein which might be responsible for its lower binding energy. The overall results indicate significant SARS-CoV-2 RdRp inhibition potential by C. longa compounds. Potent binding of the lead C. longa phytochemicals to the targeted SARS-CoV-2 and human host proteins are depicted in fig. 10.

Fig. 10: C. longa compounds targeting SARS-CoV-2 and human host proteins involved in viral entry and virulence
Author’s contributions:
Shashank Kumar acts as a co-corresponding author.
Acknowledgements:
Prem Prakash Kushwaha and Mohd Shuaib acknowledge financial support from Indian Council of Medical Research (ICMR), India in the form of ICMR-Senior Research fellowship. Shashank Kumar acknowledges University Grants Commission, India and Department of Science and Technology, India for providing financial support in the form of UGC- BSR Research Start-Up-Grant [No. F.30–372/2017 (BSR)] and DST-SERB Grant [EEQ/2016/000350] respectively. Shashank Kumar acknowledges Central University of Punjab, Bathinda, India for providing Research Seed Money Grant [GP-25]. Atul Kumar Singh and Kumari Sunita Prajapati acknowledge CSIR- India and DBT-India funding agencies for providing financial assistance in the form of Junior Research Fellowship respectively. Sanjay Gupta acknowledges support from the Department of Defense grant funds W81XWH-18-1-0618 and W81XWH-19-1-0720.
Conflict of Interests:
The authors declared no conflict of interest.
References
- Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int J Antimicrob Agents 2020;55(3):105924.
- Moriyama M, Hugentobler WJ, Iwasaki A. Seasonality of respiratory viral infections. Ann Rev Virol 2020;7:83-101.
- Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020;367(6485):1444-8.
- Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B 2020;10(5):766-88.
- Ojosnegros S, Beerenwinkel N. Models of RNA virus evolution and their roles in vaccine design. Immunome Res 2010;6(2):1-14.
- Pushpendra S, Kushwaha PP, Shashank K. Novel potent inhibitors of Plasmodium vivax dihydrofolate reductase: An in silico anti-malarial drug discovery. Indian J Pharm Educ Res 2018;52(1):122-34.
- Mishra A, Kumar S, Bhargava A, Sharma B, Pandey AK. Studies on in vitro antioxidant and antistaphylococcal activities of some important medicinal plants. Cell Mol Biol 2011;57(1):16-25.
- Sharma AK, Kumar S, Pandey AK. Ferric reducing, anti-radical and cytotoxic activities of Tinospora cordifolia stem extracts. Biochem Anal Biochem 2014;3(2):1-5.
- Lin LT, Hsu WC, Lin CC. Antiviral natural products and herbal medicines. J Tradit Complement Med 2014;4(1):24-35.
- Mani JS, Johnson JB, Steel JC, Broszczak DA, Neilsen PM, Walsh KB, et al. Natural product-derived phytochemicals as potential agents against coronaviruses: A review. Virus Res 2020;284:197989.
- Cai Y, Xu W, Gu C, Cai X, Qu D, Lu L, et al. Griffithsin with a broad-spectrum antiviral activity by binding glycans in viral glycoprotein exhibits strong synergistic effect in combination with a pan-coronavirus fusion inhibitor targeting sars-cov-2 spike s2 subunit. Virol Sin 2020;35(6):857-60.
- Keyaerts E, Vijgen L, Pannecouque C, Van Damme E, Peumans W, Egberink H, et al. Plant lectins are potent inhibitors of coronaviruses by interfering with two targets in the viral replication cycle. Antiviral Res 2007;75(3):179-87.
- Cheng PW, Ng LT, Chiang LC, Lin CC. Antiviral effects of saikosaponins on human coronavirus 229E in vitro. Clin Exp Pharmacol Physiol 2006;33(7):612-6.
- Rathore S, Mukim M, Sharma P, Devi S, Nagar JC, Khalid M. Curcumin: A review for health benefits. Int J Res Rev 2020;7(1):273-90.
- Chen TY, Chen DY, Wen HW, Ou JL, Chiou SS, Chen JM, et al. Inhibition of enveloped viruses infectivity by curcumin. PLoS One 2013;8(5):e62482.
- Mounce BC, Cesaro T, Carrau L, Vallet T, Vignuzzi M. Curcumin inhibits Zika and chikungunya virus infection by inhibiting cell binding. Antiviral Res 2017;142:148-57.
- Vajragupta O, Boonchoong P, Morris GM, Olson AJ. Active site binding modes of curcumin in HIV-1 protease and integrase. Bioorg Med Chem Lett 2005;15:3364-68.
- Kannan S, Kolandaivel P. Antiviral potential of natural compounds against influenza virus hemagglutinin. Comput Biol Chem 2017;71:207-18.
- Gupta S, Singh AK, Kushwaha PP, Prajapati KS, Shuaib M, Senapati S, et al. Identification of potential natural inhibitors of SARS-CoV2 main protease by molecular docking and simulation studies. J Biomol Struct Dyn 2021;39(12):4334-45.
- Poolsup N, Suksomboon N, Kurnianta PD, Deawjaroen K. Effects of curcumin on glycemic control and lipid profile in prediabetes and type 2 diabetes mellitus: A systematic review and meta-analysis. PLoS One 2019;14(4):e0215840.
- Petricevich VL, Abarca-Vargas R. Allamanda cathartica: A review of the phytochemistry, pharmacology, toxicology and biotechnology. Molecules 2019;24(7):1238.
- Xie XQ. Exploiting PubChem for virtual screening. Expert Opin Drug Discov 2010;5(12):1205-20.
- O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform 2011;3(1):1-4.
- Dallakyan S. PyRx-python prescription v. 0.8. The Scripps Research Institute 2008;2010.
- Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, et al. The protein data bank. Acta Crystallogr D Biol Crystallogr 2002;58(6):899-907.
- Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res 2003;31(13):3381-5.
- Xu D, Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J 2011;101(10):2525-34.
- Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 2000;302(1):205-17.
- Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 2014;42(W1):W320-4.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem 2004;25(13):1605-12.
- Tian W, Chen C, Lei X, Zhao J, Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res 2018;46(W1):W363-7.
- Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 2010;31(2):455-61.
- Laskowski RA, Swindells MB. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J Chem Inf Model 2011;51(10):2778-86.
- Li W, Wang S, Feng J, Xiao Y, Xue X, Zhang H, et al. Structure elucidation and NMR assignments for curcuminoids from the rhizomes of Curcuma longa. Magn Reson Chem 2009;47(10):902-8.
- Kita T, Imai S, Sawada H, Seto H. Isolation of dihydrocurcuminoids from cell clumps and their distribution in various parts of turmeric (Curcuma longa). Biosci Biotechnol Biochem 2009;73(5):1113-7.
- Park SY, Kim DS. Discovery of natural products from Curcuma longa that protect cells from beta-amyloid insult: A drug discovery effort against Alzheimer's disease. J Nat Prod 2002;65(9):1227-31.
- Gokaraju GR, Gokaraju RR, Gottumukkala VS, Somepalli V, inventors; Laila Impex, assignee. Process for producing enriched fractions of tetrahydroxycurcumin and tetrahydrotetrahydroxy-curcumin from the extracts of Curcuma longa. United States patent 2013;8:1-6.
- Chen JJ, Tsai CS, Hwang TL, Shieh PC, Chen JF, Sung PJ. Sesquiterpenes from the rhizome of Curcuma longa with inhibitory activity on superoxide generation and elastase release by neutrophils. Food Chem 2010;119(3):974-80.
- Yannai S, editor. Dictionary of food compounds with CD-ROM. Crc Press; 2012.
- Matsuo Y, Mimaki Y. α-Santalol derivatives from Santalum album and their cytotoxic activities. Phytochem 2012;77:304-11.
- Thaikert R, Paisooksantivatana Y. Variation of total curcuminoids content, antioxidant activity and genetic diversity in turmeric (Curcuma longa L.) collections. Agri Nat Resour 2009;43(3):507-18.
- Lim TK. Edible medicinal and non-medicinal plants. Dordrecht, The Netherlands: Springer; 2016;12:371-88.
- Li W, Feng JT, Xiao YS, Wang YQ, Xue XY, Liang XM. Three novel terpenoids from the rhizomes of Curcuma longa. J Asian Nat Prod Res 2009;11(6):569-75.
- Wang LY, Zhang M, Zhang CF, Wang ZT. Diaryl derivatives from the root tuber of Curcuma longa. Biochem Syst Ecol 2008;5(36):476-80.
- Duraisankar M, Ravindran AD. Identification of Curcuma longa rhizomes by physicochemical and TLC fingerprint analysis. Int J Pharm Tech Res 2015;8(6):198-205.
- Saladini M, Lazzari S, Pignedoli F, Rosa R, Spagnolo F, Ferrari E. New Synthetic Glucosyl-Curcuminoids and their 1 H and 13 C NMR Characterization, from Curcuma longa L. Plant Foods Hum Nutr 2009;64(3):224-9.
- Esparan V, Krings U, Struch M, Berger RG. A three-enzyme-system to degrade curcumin to natural vanillin. Molecules 2015;20(4):6640-53.
- Senapati S, Kumar S, Singh AK, Banerjee P, Bhagavatula S. Assessment of risk conferred by coding and regulatory variations of TMPRSS2 and CD26 in susceptibility to SARS-CoV-2 infection in human. J Genet 2020;99:1-5.
- Nelson KM, Dahlin JL, Bisson J, Graham J, Pauli GF, Walters MA. The essential medicinal chemistry of curcumin: Miniperspective. J Med Chem 2017;60(5):1620-37.
- Zhou H, S Beevers C, Huang S. The targets of curcumin. Curr Drug Target 2011;12(3):332-47.
- Goel A, Kunnumakkara AB, Aggarwal BB. Curcumin as “Curecumin”: From kitchen to clinic. Biochem Pharmacol 2008;75(4):787-809.
- Gupta SC, Patchva S, Koh W, Aggarwal BB. Discovery of curcumin, a component of golden spice and its miraculous biological activities. Clin Exp Pharmacol Physiol 2012;39(3):283-99.
- Patwardhan M, Morgan MT, Dia V, D'Souza DH. Heat sensitization of hepatitis A virus and Tulane virus using grape seed extract, gingerol and curcumin. Food Microbiol 2020;90:103461.
- Lu G, Hu Y, Wang Q, Qi J, Gao F, Li Y, et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 2013;500(7461):227-31.
- Shen LW, Mao HJ, Wu YL, Tanaka Y, Zhang W. TMPRSS2: A potential target for treatment of influenza virus and coronavirus infections. Biochimie 2017;142:1-10.
- Dimitrov DS. Virus entry: Molecular mechanisms and biomedical applications. Nat Rev Microbiol 2004;2(2):109-22.
- Lin LT, Chen TY, Lin SC, Chung CY, Lin TC, Wang GH, et al. Broad-spectrum antiviral activity of chebulagic acid and punicalagin against viruses that use glycosaminoglycans for entry. BMC Microbiol 2013;13(1):1-5.
- Ciesek S, von Hahn T, Colpitts CC, Schang LM, Friesland M, Steinmann J, et al. The green tea polyphenol, epigallocatechin?3?gallate, inhibits hepatitis C virus entry. Hepatol 2011;54(6):1947-55.
- Biftu T, Sinha-Roy R, Chen P, Qian X, Feng D, Kuethe JT, et al. Omarigliptin (MK-3102): a novel long-acting DPP-4 inhibitor for once-weekly treatment of type 2 diabetes. J Med Chem 2014;57:3205-12.
- Iheagwam FN, Ogunlana OO, Chinedu SN. Model optimization and in silico analysis of potential dipeptidyl peptidase IV antagonists from GC-MS identified compounds in Nauclea latifolia leaf extracts. Int J Mol Sci 2019;20(23):5913.
- Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020;181(2):281-92.
- Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect 2020;9(1):221-36.
- Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun 2020;11(1):1620.
- Tai W, He L, Zhang X, Pu J, Voronin D, Jiang S, et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol 2020;17(6):613-20.
- Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020;367(6483):1260-3.
- Tong TR. Drug targets in severe acute respiratory syndrome (SARS) virus and other coronavirus infections. Infect Disord Drug Targets 2009;9(2):223-45.