- *Corresponding Author:
- A. Saha
Department of Chemical Technology, University of Calcutta, 92 A. P. C. Road, Kolkata–700 009, India.
E-mail: achintya_saha@yahoo.com
| Date of Submission | 29 December 2005 |
| Date of Revision | 15 June 2006 |
| Date of Acceptance | 11 February 2007 |
| Indian J Pharm Sci, 2007, 69 (1): 125-129 |
Abstract
Broad range of structurally diverse alkylphenols has been found to be considerably potential estrogenic agents in combating estrogen-linked pathologies, but their mechanism of action in mimicking responses of endogenous hormones is still to be understood. The present work explores pharmacophore signals of some varied alkylphenols and predicts estrogenic activities through generated linear relations implementing theoretical molecular modeling techniques. The binding affinity to estrogen receptor of alkylphenols has been modeled investigating large data set of whole molecular and atomic descriptors. Univariate and multivariate relationships were estimated using correlation analysis and statistical significance of the generated relations assessed. The predictive ability of the generated models was further verified using 'Leave-One-Out' cross validation. The relationships with molecular properties could be developed with a maximum correlation exceeding 94%, with explained variance as high as 87% and cross-validated variances >0.8. It was inferred that increased molecular bulk, enhanced molecular ionization potential, presence of electron donating groups in para position and branched chain terminal atoms might have influence on binding affinity to the receptor.
Synthetic endocrine-disrupting chemicals (EDCs) bear the possibility to interfere in the endocrine system by mimicking endogenous hormones such as estrogens and androgens [1]. Among these classes, diethylstilbestrol (DES), dichlorodiphenyltrichloroethanes/ethylenes (DDTs), polychlorinated biphenyls (PCBs), alkylphenols, phthalates and parabens have been found to be estrogenic, that display a broad range of structural diversity [2]. Xenoestrogens [3], known as the environmental estrogens are comprised of widely varied structures, including isoflavonoids, phytoestrogens such as genistein, the mycotoxin-zeranol and industrial chemicals such as mbisphenol A. Different xenoestrogens are also known to interact with the estrogen receptor-α subtype [4].
Recently alkylphenols have been explored as prospective agents in combating estrogen-linked pathologies, such as antiproliferative agents in breast cancer treatment [5]. However, their mechanism of action in mimicking responses of endogenous hormones is still in dark [6]. Estrogenic activity is distinctive in that it does not require a steroidal structural configuration, as do the other sex hormones. Molecules with a distance limitation of 10.3-12.1 Å between the oxygen atoms of the hydroxyl groups on a large skeleton have optimal estrogenic activity [7]. Nevertheless, certain plant flavonoids with only one phenolic hydroxyl group have also been investigated to be appreciably estrogenic [6,8]. Computer-based tools have enabled to develop quantitative structure-activity relationship (QSAR) models for identifying steric and electronic properties of a molecule for estrogenic activity [9-11]. Though a number of chemical classes are known to be estrogenic and many techniques continue to be in implementation for defining pharmacophore signal for these, still little thorough structural evaluation has been presented in comparison to the endogenous estrogen ligands [6]. A linear correlation of 91% was earlier established with hydrophobicities of alkylphenols (n=24) for binding with the estrogen receptor (ER), where quantum of Log P was found to be directly proportional to the binding affinity [6]. This was partly substantiated by another report [12] that established nearly 65% correlation with hydrophobicity factor and significant (r2 =0.92) whole molecular contribution (such as molecular volume and ionization potential) towards estrogenic activities of structurally diverse phenolic compounds (n=18) including steroidal moieties, that are known to possess high molecular volume and ionization potential, leading to increased binding affinity [7]. It has also been explored that phenolic OH groups, m-substitutions in phenyl rings and bridging alkyl moieties between phenyl segments of bisphenol A derivatives (n=20) might be essential for estrogenic activities [13] and presence of -Cl as the m-substituent might greatly influence the activity towards selectivity for the estrogen receptor-α subtype [14]. In comparison, the present work explores the probable supplementary structural requirements of a diverse and fairly large set of estrogenic non-steroidal alkylphenols by investigating bulk, steric and electronic properties of the compounds.
A large variety of electronic and steric molecular descriptors have been used to encode electronic features in QSAR and QSPR studies [15]. Molar refractivity (MR), a function of steric influence of a molecule is a measure of molecule’s susceptibility to become polarized. It is mathematically defined [16] as, 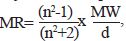 where MW is the molecular weight, d, density and n the refractive index. The term MW/d defines a volume, while the (n2-1)/(n2 +2) term provides a correction factor that defines the substituent polarizability. Reactivity indices are usually categorized as either electrophillic or nucleophillic, depending on whether the reaction of interest involves electrophillic or nucleophillic attack. The simplest of such descriptors are EHOMO and ELUMO, the energies of the highest occupied and lowest unoccupied molecular orbitals, respectively. The HOMO energy is related to the ionization potential of the molecule, while the LUMO energy is related to the electron affinity. The magnitudes of these quantities are the measure of the overall susceptibility of the molecule to losing a pair of electrons to an electrophile or accepting a pair of electrons from a nucleophile [15].
where MW is the molecular weight, d, density and n the refractive index. The term MW/d defines a volume, while the (n2-1)/(n2 +2) term provides a correction factor that defines the substituent polarizability. Reactivity indices are usually categorized as either electrophillic or nucleophillic, depending on whether the reaction of interest involves electrophillic or nucleophillic attack. The simplest of such descriptors are EHOMO and ELUMO, the energies of the highest occupied and lowest unoccupied molecular orbitals, respectively. The HOMO energy is related to the ionization potential of the molecule, while the LUMO energy is related to the electron affinity. The magnitudes of these quantities are the measure of the overall susceptibility of the molecule to losing a pair of electrons to an electrophile or accepting a pair of electrons from a nucleophile [15].
The attribute of atoms or groups in a molecule that bind with a receptor or an enzyme is mostly electronic in nature. An atom in a molecule is part of a field of information relating to electronic influences [17,18]. Atomic charges are important descriptors for defining electronic contribution of atoms in a molecule [19].
In the present study a set of 28 molecules (Table 1) belonging to different alkylphenols exhibiting estrogenic activity [6] were selected. Biological activity (binding affinity) is expressed in terms of negative logarithm of relative binding affinity (pRBA=-Log RBA) to the estrogen receptor. For a particular molecule, the RBA to the estrogen receptor is defined as 100 times the ratio of IC50 of 17ß estradiol to the IC50 of the molecule (RBA of 17ß estradiol= 100).

Table 1: Observed, calculated and predicted biological activities (relative binding affinity to estrogen receptor) of alkylphenols
The descriptors were calculated with CS Chem3D 5.0 [20,21]. The molecules were energy minimized to a minimum RMS gradient of 0.1 in CS MOPAC 5.0 using AM1 method with Close shell (restricted) wave function. The charge functions were calculated using Wang-Ford approach [21]. QSAR model origination was accomplished by correlation analysis. Statistical analyses were performed using standard and forward stepwise multiple regression [22]. All the data were statistically analyzed through computation of: r or R (correlation coefficient), EV (explained variance), F (variance ratio) with df (degrees of freedom), s (standard error of estimate), AVRES (average of absolute values of residuals). Leave-One-Out cross-validation [23] was performed that generated PRESS (predictive residual sum of squares), SDEP (standard deviation of error of predictions), Presav (average of absolute value of predicted residuals) and Q2 (Cross-validated variance).
The best univariate relationship for binding affinity to the ER with 28 nos. of structurally diverse alkylphenols [6] was found to be:
pRBA = 6.175(±0.279) - 0.018(±0.001) MWr = 0.925, r2 = 0.855, EV = 84.992%, F (df) = 153.910 (1, 26),s = 0.338, AVRES = 0.241, PRESS = 3.297, SDEP = 0.343, Presav = 0.256, Q2 = 0.839, n = 28
Eqn. 1 explained the importance of molecular bulk interms of molecular weight (MW) of alkylphenols forbinding affinity to the ER. But when multivariateapproach was considered for elucidating the possibility ofany pooled involvement of molecular propertiesaccountable for binding affinity to the ER involving all the 28 compounds, a new relationship was developed with 87.14% explained variance.
pRBA = 4.273(±0.849) - 0.057(±0.005) MR0.186(±0.062)EHOMO+ 0.660(±0.291) Ch4 - 0.160(±0.055) Nt, R = 0.944, R2 = 0.890, EV = 87.138%, F (df) = 46.731 (4, 23), s = 0.313, AVRES = 0.220, PRESS = 3.318, SDEP = 0.344, Presav = 0.269, Q2 = 0.838, n = 28 (2) where Ch4 is the charge function of C4 and Nt is the no. of free terminal atoms (excluding H) that indicates the degree of substitutions in the phenolic ring. For e.g., in compound 25 (Table 1), Nt=5 (one oxygen atom of OH and four chlorine atoms in the phenyl ring side chain). The 95% confidence intervals are shown in parentheses and the F- values are significant at 99% confidence level. The independent variables used in Eqn. 2 are not intercorrelated, |r|≤0.5 (Table 2). The calculated and predicted activities obtained from the equations (1) and (2) are delineated in Table 1 and the graphical representation of observed versus predicted values of Eqn. 2 is depicted in fig. 1.
| Variables | r | |||
|---|---|---|---|---|
| MR | EHOMO | Ch4 | Nt | |
| MR | 1.000 | 0.249 | 0.112 | 0.064 |
| EHOMO | 1.000 | 0.026 | 0.092 | |
| Ch4 | 1.000 | 0.238 | ||
| Nt | 1.000 | |||
Table 2: Intercorrelation (r) matrix of independent variables used in eqn. 2

Figure 1: Relative binding affinity (n= 28) to the estrogen receptor. Estrogen receptor binding affinity of alkylphenols from Eqn. 2
The best univariate relation developed (92.5% correlation) with MW (Eqn. 1) that explained 84.992% variance in observed magnitude with 83.9% cross-validated variance. The model thus shows importance of molecular bulk in binding to the ER. Hu and Aizawa [12] have also substantiated this fact in terms of molecular volume (r2= 0.89), where the bulk parameter was also found to be a positive contributor towards activity The coefficient of MW in the equations isnegative; hence less bulky molecules will exhibitlesser binding affinity to the ER. Compound 25 withthe highest molecular weight (307.991) in the seriesexhibits highest binding while Compound 9 with theleast molecular weight (122.168) exhibits the leastbinding affinity. Eqn. 2 explained 87.138% variance inobserved binding affinity with 83.836% cross-validatedvariance. This model could explain importance of molar refractivity (steric influence and bulkiness), ionization potential of the molecules (in terms of EHOMO), electron density of C4 atom and branching effects of the molecules towards binding. In the model, MR has negative coefficient, hence substitutions that tend to decrease the steric influence or a decrease in overall molecular size will have negative impact on binding affinity to the ER. Compounds 9 and 10 with minimum size have the least binding affinity. The estrogenic activities of alkylphenols are largely dependent on the alkyl chain length at the para position. Generally, the activity increases with the increase in chain length [6,13,24]. The factor Nt has also negative coefficient, so compounds with more number of terminal atoms (excluding H) should also result in increased binding affinity. In general compounds with more branched side chain substitutions resulting in more Nt (Compounds 6, 19 and 25) has increased binding affinities in comparison to rest of the series. Importance of free terminal atoms in binding affinity to the estrogen receptor has been demonstrated earlier with triphenylacrylonitriles as prototype molecules [25].
The factor EHOMO has negative coefficient, so decrease in ionization potential of the molecules will also result in lesser binding to the ER [12]. Furthermore as Ch has positive coefficient, therefore more electron density on C4 is desirable for more binding affinity. Compounds 2 and 3 with long chain alkyl groups exhibit favorable + I effect for enhancing electron density on C4 and therefore have comparatively higher activity.
The present in silico study could account for structural requirements of alkylphenols for ER binding affinity. The models generated with cross-validated variances >80% suggests their consistent predictive ability [26]. This study supports that increased molecular bulk, i.e., steric impact, enhanced molecular ionization potential, presence of electron donating groups in para position of alkylphenols and more branched chains could impart more active compounds
Acknowledgements
Financial assistance received from AICTE, New Delhi under Research Promotion Schemes is gratefully acknowledged. One of the authors, S. Mukherjee also acknowledges CSIR, New Delhi for grant of Senior Research Fellowship.
References
- Kavlock, R.J., Daston, G.P., DeRosa, C., Fenner-Crisp, P., Gray, L.E., Kaattari, S., Lucier, G., Luster, M., Mac, M.J., Maczka, C., Miller, R., Moore, J., Rolland, R., Scott, G., Sheehan, D.M., Sinks, T. and Tilson, H.A., Environ. Health Perspect ., 1996, 104, 715.
- Katzenellenbogen, J.A., Environ. Health Perspect ., 1995, 103, 99.
- Adlercreutz, H., Environ. Health Perspect ., 1995, 103, 103.
- Wozniak, A.L., Bulayeva, N.N. and Watson, C.S., Environ. Health Perspect ., 2005, 113, 431.
- Vessieres, A., Top, S., Pigeon, P., Hillard, E., Boubeker, L., Spera, D. and Jaouen, G., J. Med. Chem ., 2005, 48, 3937.
- Fang, H., Tong, W., Shi, L. M., Blair, R., Perkins, R., Branham, W., Hass, B.S., Xie, Q., Dial, S.L., Moland, C.L. and Sheehan, D.M., Chem. Res. Toxicol ., 2001, 14, 280.
- Brueggemmeier, R.W., Miller, D.D. and Dalton, J.T., In; Williams, D.A. and Lemke, T.K., Eds., Foye's Principles of Medicinal Chemistry, 5th Edn., Lippincott Williams & Wilkins, Philadelphia, 2002, 692.
- Branham, W.S., Dial, S.L., Moland, C.L., Hass, B.S., Blair, R.M., Fang, H., Shi, L., Tong, W., Perkins, R.G. and Sheehan, D.M., J. Nutr ., 2002, 132, 658.
- Sadler, B.R., Cho, S.J., Ishaq, K.S., Chae, K. and Korach, K.S., J. Med. Chem ., 1998, 41, 2261.
- Gantchev, T.G., Ali, H. and van Lier, J.E., J. Med. Chem ., 1994, 37, 4164.
- Waller, C.L., Minor, D.L. and McKinney, J.D., Environ. Health Perspect ., 1995, 103, 702.
- Hu, J.Y. and Aizawa, T., Water Res ., 2003, 37, 1213.
- Kitamura, S., Suzuki, T., Sanoh, S., Kohta, R., Jinno, N., Sugihara, K., Yoshihara, S., Fujimoto, N., Watanabe, H. and Ohta, S., Toxicol. Sci ., 2005, 84, 249.
- Takemura, H., Ma, J., Sayama, K., Terao, Y., Zhu, B.T. and Shimoi, K., Toxicology , 2005, 207, 215.
- Jurs, P.C., Dixon, S.L. and Egolf, L.M., In; Waterbeemd, H.V de., Eds., Methods and Principles in Medicinal Chemistry, Vol. II, VCH Verlagsgesellschaft mbh, Weinheim, 1995, 15.
- Atkins, P.W., In; Physical Chemistry, Freeman, New York, 1982, 92.
- Kier, L.B. and Hall, L.H., Pharm. Res ., 1990, 7, 801.
- Hall, L.H., Mohney, B. and Kier, L.B., Quant. Struct-Act. Relat., 1991, 10, 43.
- House, J. E., In; Fundamentals of Quantum Chemistry, Academic Press, California, 2005, 275.
- Ghose, A.K. and Crippen, G.M., J. Chem. Inf. Comput. Sci ., 1987, 27, 21.
- Brand name of Cambridge Soft, Corp. , Cambridge (USA).
- Statistica 5.0, StatSoft, inc. , Tulsa (USA).
- Wold, S. and Eriksson, L., In; Waterbeemd, H. V de., Eds., Chemometric Methods in Molecular Design (Methods and Principles in Medicinal Chemistry), Vol. II, VCH Verlagsgesellschaft mbh, Weinheim, 1995, 312.
- Routledge, E. J. and Sumpter, J. P., J. Biol. Chem . , 1997, 272, 3280.
- Mukherjee, S., Mukherjee, A. and Saha, A., Biol. Pharm. Bull ., 2005, 28, 154.
- Clementi, S. and Wold, S., In; Waterbeemd, H. V de., Eds., Chemometric Methods in Molecular Design (Methods and Principles in Medicinal Chemistry), Vol. II, VCH Verlagsgesellschaft mbh, Weinheim, 1995, 324.