- *Corresponding Author:
- O. Inal
Department of Pharmaceutical Technology, Faculty of Pharmacy, University of Ankara, Tandoğan Ankara-06100, Turkey
E-mail: uzmecz@yahoo.com
| Date of Submission | 23 April 2013 |
| Date of Revision | 12 October 2013 |
| Date of Acceptance | 19 October 2013 |
| Indian J Pharm Sci 2013;75(6):700-706 |
Abstract
Thermoreversible gel of meloxicam, efficient for the treatment of joint diseases, was aimed to prepare for night application available for chronotherapy in this study. Poloxamer 407 and 188 polymers were used at 20-30% w/w as a vehicle in combination with different additives (polyvinylmethylether maleic anhydride copolymer, hydroxypropyl methylcellulose, polyethylene glycol 400, dimethyl sulfoxide, sodium chloride). Characterisation of prepared gels was evaluated by viscosity and texture analysis, and the effect of formulation variables on the gel formulations were evaluated by in vitro drug release and erosion studies. Between the investigated gel bases, Poloxamer 407-hydroxypropyl methylcellulose gel was found to be ideal due to its gel strength (1.560±0.0135 N), viscosity (312.3±2.06 cP) and release characteristics. These promising results could be encouraging for further studies to make it an alternative to commercial dosage forms.
Keywords
Chronotherapy, hydrogel, meloxicam, thermoreversible, transdermal delivery
Sustained transdermal therapy has gained importance in the past decades not only for local but also for systemic drug delivery especially for drugs have first-pass metabolism and gastrointestinal disturbance. Since the nonsteroidal antiinflammatory drugs (NSAIDs) are administered for long-term therapy, it is desirable to reduce the dermal reactions [1] such as skin irritation due to direct contact between drug and skin. It has been reported that meloxicam can be applied to the skin and mucosa due to its lower tissue toxicity than the other NSAID, which make it a good alternative for patients [2] suffering from joint diseases such as rheumatoid arthritis and osteoarthritis. However, photosensitisation can be occurred by ultraviolet (UV) exposure of skin due to applied a transdermal NSAID formulation in daytime [3,4], hereby it should be better to apply such dosage forms during night time thus enhance the bioavailability due to circadian rhythm for nocturnal pain or effectively control morning stiffness and pain [5]. In the case of rheumatoid arthritis, patients wake up with pain in the mornings; therefore application of a NSAID on the aching part of body during night can be a solution due to effects on the rhythmic patterns in pain level and their chronotherapy.
Polyethylen oxide-polypropylene oxide-Polyethylen oxide (PEO-PPO-PEO) triblock copolymers known as poloxamers (such as Pluronic®, Lutrol®) with relatively low toxicity and ability to form clear gels in aqueous media, have been widely used in pharmaceutical field [6-14]. The unique characteristic of this copolymer is reverse thermal gelation behaviour, which occurs at concentrations above 20% w/w. Highly concentrated solutions of copolymers is in solution form below the critical micellization temperature (CMT), which forms soft gels above the CMT. In practice, this resulted with a gelation in a range between room or body temperature, due to concentration of polymer and/or existence of excipients. Thus, polymeric solutions could be easily prepared at refrigerator temperature (4-5°) and owing to be ease of preparation, exhibiting reverse thermal gelation and good drug-release characteristics, copolymer gels become good alternatives for topical delivery [6,7]. Additionally, they form swellable hydrogels, which give more emollient effect than conventional ointments due to their high water content [7] that have a positive effect on absorption of drugs. Gel properties such as critical micelle concentration (CMC), CMT, gel strength (hardness), adhesiveness of these copolymers can be modified by some excipients like salts (NaCl, Na2SO4, NaH2PO4 and MgSO4, CaCl2), hydroxylated compounds (polyethylene glycol, glycerin and propylene glycol) or various polymers such as carbopol, hydroxypropyl methylcellulose (HPMC), polyvinylmethylmethacrylate and solvents like dimethyl sulphoxide (DMSO). Mechanical properties of poloxamer gels can also be affected by the physicochemical properties of the incorporated drug [7,9-14].
In this study, it was aimed to prepare a poloxamer-based thermoreversible transdermal dosage form of meloxicam, which should be applied during night time as a gel or a patch thus enhance the bioavailability due to circadian rhythm for rheumatoid arthritis. Gel strength, viscosity and drug release characteristics in the light of the gelation temperature, viscosity, texture, in vitro dissolution and gel erosion studies were performed on investigated formulations.
Materials and Methods
Poloxamer 407 (Pluronic F-127®, Sigma, USA; F127) and Poloxamer 188 (Pluronic F-68®, Sigma, USA; F68), Polyvinyl maleic acid copolymer-PVM/MA copolymer (Gantrez® S-97 BF; S-97) was a gift from ISP Chem, Germany. Hydroxypropyl methylcellulose (Methocel K100 PRM LV, Dow Chemical Company, Germany; K100LV), Polyethylene glycol 400 (PEG 400, Merck), DMSO (Merck), Sodium chloride (NaCl, Merck), Meloxicam was a gift from Fargem, Turkey.
Preparation of transdermal gels
Transdermal gel formulations were prepared as “cold method” described by Schmolka [15]. Briefly, weighed amount of copolymer was slowly added to cold water then stored in refrigerator (4-5°) for 48 h and gently mixed (100 rpm) in cold environment for 10-15 min at every 12 h periods, to ensure complete dissolution. Clear and viscous gel or solution formed eventually [6]. Drug and other excipients including formulations were prepared by adding (weight percentage) all other ingredients to cold water before the polymers.
Characterisation
Gelation temperatures (Tgel) were measured on a thermostatic magnetic stirrer (at 100 rpm) until the in situ gel occurs by micellization (n=3). Viscosity measurements were performed by using a rotational viscometer (Brookfield DVII) and TF spindle at 10 rpm, 37° (n=3).
Texture profile analyses (TPA) were carried out with TA.XT Plus Texture Analyser (Stable Micro Systems, UK) in TPA mode. The formulations were transferred into McCartney type bottles (of identical dimensions with a fixed height) then the samples equilibrated at room temperature for 1 h. An analytical probe (Perspex, P/0.5) was compressed twice into each sample to a depth of 10 mm at a rate of 2 mm/s (both for pretest, test and posttest speeds) allowing a delay period of 5 s between the two compressions. Three replicates were performed at room temperature with a fresh sample in each case. From the resultant force-time plot of TPA graph, mechanical parameters such as hardness, compressibility and adhesiveness were defined [16,17].
In vitro drug release studies
In vitro drug release studies were performed with a membrane-less method [18]. Briefly, 1 g of polymeric solution (+4°) was weighed in a 10 ml vial (1.5 cm in diameter), which was cooled at +4° before and gelled at 37° in an oven (gel had a smooth surface without any bubbles). Then 1 ml of phosphate buffered saline (PBS) buffer (pH 7.4, 37°) was layered on the gel in an incubator (MaxQ 4450, ThermoScientific). Samples were incubated (50 rpm, 37°) for 6 h. At predetermined time intervals (0, 1, 2, 3, 4, 5 and 6 h), the entire release medium was taken out and renewed with fresh buffer (n=3). Samples were measured spectrophotometrically at 365 nm (Shimadzu 1204, Japan). Analytical validation of the spectrophotometric method was done with precision, accuracy, linearity and range.
In vitro gel erosion studies
In vitro gel erosions (% w/w) were studied during in vitro release studies, as described earlier [18], parallel to sampling the vials were weighed after the removal of the entire release medium (the vials were completely dried and kept at 37° before weighing) at each time point and the differences in weights of vials give the amount of gel dissolved. The erosion profiles of formulations were then obtained from the cumulative weight of each gel dissolved versus time.
Data analysis of drug release and gel erosion studies
The data of in vitro drug release studies were analysed according to zero order, first order, Higuchi models and Korsmeyer–Peppas release kinetics. In vitro drug release data was plotted against in vitro gel erosion data and the obtained r2 values were used to evaluate the relation between gel erosion and the release of meloxicam.
Results and Discussion
In this study, Pluronic F127 (F127) in situ gels, either alone or combined with Pluronic F68 (F68), were modified with different excipients in order to prepare a suitable gel formulation for 6 h of durability. Codes and contents of formulations are given in Table 1 and the mechanical properties derived from force-time curve of TPA graphs and CMT values are given in Table 2.
| Code | Polymer | F127:F68 | MLX | S-97 | K100LV | DMSO | PEG400 | NaCl |
|---|---|---|---|---|---|---|---|---|
| F1 | 30 | 1:00 | - | - | - | - | - | - |
| F2 | 30 | 3:01 | - | - | - | - | - | - |
| F3 | 30 | 4:01 | - | - | - | - | - | - |
| F4 | 30 | 5:01 | - | - | - | - | - | - |
| F5 | 26 | 1:00 | - | - | - | - | - | - |
| F6 | 26 | 3:01 | - | - | - | - | - | - |
| F7 | 26 | 4:01 | - | - | - | - | - | - |
| F8 | 26 | 5:01 | - | - | - | - | - | - |
| F9 | 20 | 1:00 | - | - | - | - | - | - |
| F10 | 20 | 3:01 | - | - | - | - | - | - |
| F11 | 20 | 4:01 | - | - | - | - | - | - |
| F12 | 20 | 5:01 | - | - | - | - | - | - |
| F1A | 30 | 1:00 | 0.1 | - | - | - | - | - |
| F4A | 30 | 5:01 | 0.1 | - | - | - | - | - |
| F5A | 26 | 1:00 | 0.1 | - | - | - | - | - |
| F8A | 26 | 5:01 | 0.1 | - | - | - | - | - |
| F9A | 20 | 1:00 | 0.1 | - | - | - | - | - |
| F5B | 26 | 1:00 | 0.1 | 0.5 | - | - | - | - |
| F5C | 26 | 1:00 | 0.1 | - | 5 | - | - | - |
| F5D | 26 | 1:00 | 0.1 | - | - | 5 | - | - |
| F5E | 26 | 1:00 | 0.1 | - | - | - | 5 | - |
| F5F | 26 | 1:00 | 0.1 | - | - | - | - | 1 |
Contents are given as percent w/w, DMSO=dimethyl sulphoxide, MLX=meloxicam, PEG=polyethylene glycol, NaCl=sodium chloride
Table 1: Contents Of Gel Bases And Transdermal Gel Formulations
| Code | Tgel (°) | ηx1000 (cP) | Gel Strength (N) | Compressibility (Ns) | Adhesiveness (Ns) |
|---|---|---|---|---|---|
| F1 | 15.0±1.0 | 317.7±7.0 | 1.483±0.0543 | 4.840±0.1486 | 4.053±0.3845 |
| F2 | 26.0±2.7 | 316.3±7.2 | 0.465±0.0053 | 1.731±0.0439 | 1.526±0.0158 |
| F3 | 19.0±1.0 | 411.0±7.0 | 0.648±0.0121 | 2.370±0.0474 | 2.104±0.0320 |
| F4 | 21.0±1.0 | 373.7±9.7 | 0.852±0.0112 | 2.945±0.1102 | 2.740±0.0535 |
| F5 | 19.0±1.7 | 285.6±2.5 | 0.910±0.0060 | 3.018±0.0249 | 2.505±0.1560 |
| F6 | 32.6±0.6 | 225.6±8.1 | 0.282±0.0108 | 1.055±0.0477 | 0.797±0.0066 |
| F7 | 27.0±1.0 | 155.7±0.6 | 0.455±0.0101 | 1.457±0.0537 | 1.518±0.1609 |
| F8 | 25.6±0.6 | 203.0±8.9 | 0.627±0.0085 | 1.990±0.0830 | 1.813±0.0593 |
| F9 | 23.7±1.5 | 287.4±3.1 | 0.465±0.0493 | 1.471±0.0737 | 1.207±0.1594 |
| F10 | 45.6±1.0 | 3.130±0.2 | * | * | * |
| F11 | 39.0±1.5 | 46.8±0.6 | * | * | * |
| F12 | 37.7±1.5 | 27.7±0.6 | * | * | * |
| F5A | 18.8±0.4 | 272.7±2.05 | 0.860±0.0263 | 3.040±0.0796 | 2.627±0.1772 |
| F5B | 14.3±0.5 | 253.0±4.10 | 0.926±0.0186 | 3.141±0.0581 | 2.769±0.0549 |
| F5C | 17.0±0.1 | 312.3±2.06 | 1.560±0.0135 | 4.773±0.1225 | 4.204±0.1653 |
| F5D | 14.1±0.5 | 350.0±4.00 | 1.168±0.0165 | 3.799±0.1084 | 3.384±0.1018 |
| F5E | 14.5±0.5 | 419.1±9.50 | 1.413±0.0354 | 4.276±0.5408 | 3.805±0.5285 |
| F5F | 16.2±0.2 | 545.2±9.07 | 1.081±0.0140 | 3.724±0.0722 | 3.359±0.0971 |
η: Viscosity, (*) solution, Ns=not significant
Table 2: Physical Properties Of Gel Bases And Transdermal Gel Formulations
In the design of gel bases, F68 was used for increasing the low CMT values of F127 gels. F127 alone, had a CMT value under room temperature, thus it is combined with different ratios of F68 (formulations F2-F4, F6-F8, F10-F12). Even though presence of F68 provided a CMT value near room temperature, lower total polymer ratio (≤20%) resulted with a very high CMT (above body temperature) and very low gel hardness with this combination (F10-F12). As a general trend, F68 caused to form weaker gel bases while the high concentration of F127 polymer (F1, 30% w/w) gave the hardest gel and the mechanical properties of formulations with low concentration (≤20% w/w, F10-F12) was not able to be measured at ambient temperature. This was attributed to the short chain and low PPO/PEO molar ratio of F68 polymer tends to an increase in CMT and CMC of the gels [19,20] (Table 2).
Between the gel bases (F1-F12) given in Table 1; F5, due to being a single polymer with moderate gel properties among the prepared ones, was chosen as a model base to evaluate the effect of S-97, K100LV, DMSO, PEG 400 and NaCl with presence of meloxicam (recoded with B, C, D, E, F in Table 1) on mechanical and release properties. Also; F1, F4, F5, F8 and F9 were recomposed with meloxicam (re-coded with A in Table 1) in order to see the effect of polymer type, concentration and additives on in vitro drug release and gel erosion.
Mechanical properties of poloxamer gels can also be affected by the physicochemical properties of incorporated drug [13]. Yong et al. indicated that poorly water soluble diclofenac sodium can be solubilised in aqueous medium by poloxamers, acted like a salt and had markedly increasing effect on poloxamer gel’s CMT [13]. Thus, effect of poorly water soluble meloxicam on texture properties was investigated on F5A, due to this formulation’s suitable gel hardness, which could be available to achieve meloxicam release for 6 h (fig.1; F5A). There has not been any significant difference obtained with Tgel and mechanical properties between F5 and F5A (Table 2) and it was concluded that meloxicam was not significantly effective on the abovementioned properties, thus mechanical properties of F1A, F4A, F8A and F9A formulations were not studied further.

Fig 1: Effect of formulation variable on meloxicam release.
(a) Effect of polymer type and concentration: F1A 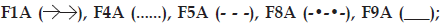 (b) Effect of excipients on in vitro
meloxicam release from transdermal formulations:
(b) Effect of excipients on in vitro
meloxicam release from transdermal formulations: 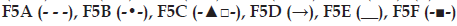 .
.
Polymers, S-97 and K100LV; cosolvents, DMSO and PEG 400; and salt, NaCl were used for modifying gel texture in order to investigate their effect on the release versus gel erosion. As seen in Table 2, all the additives significantly increased the gel hardness, except S-97. S-97 is a very tacky polymer, which is usually used in bioadhesive gel formulations [21] and combination of 4% w/w S-97 with poloxamer and HPMC has significantly increased the detachment force of the formulations prepared by Dhiman et al. [21]. Solutions of S-97 found to be highly viscous at pH 6.8 because of ionisation of carboxylic acid groups. However, in our study, we did not obtain such high viscosity results, probably because of using a relatively low concentration of this polymer. Higher concentrations such as 4-5% w/w could not homogenously formulate with the addition of poloxamers over a 20% w/w concentration. In the evaluation of F5 gel bases, only meloxicam did not change the CMT value; however, other additives significantly decreased the gelling temperatures (Table 2).
In evaluating the effect of poloxamer type and concentration on in vitro release of meloxicam; presence of F68 polymer increased the release of meloxicam (F4A and F8A) probably due to the change in the ratio of PPO/PEO units in the polymer (fig. 1a). Comparably shorter chain and low PPO/PEO molar ratio of hydrophilic F68 tends to disrupt the hydration shells around the hydrophobic portion of F127 molecules, which resulted as high degree of water molecules around the PPO units. During gelation those ordered water molecules had to be squeezed out into the bulk solution. Therefore, an increase in temperature required to promote the hydroscopic interaction between poloxamer micelles [21]. Thus, gel prepared with F68 has more tendencies to erode. As seen in fig. 2a, formulations including F127:F68 combination (F4A and F8A) showed higher erosion profiles than the others probably due to the decrease in PPO/PEO molar ratio of polymer in the gel. Thus, existence of F68 resulted with higher erosion profiles.

Fig 2: Effect of formulation variable on gel erosion.
(a) Effect of polymer type and concentration: 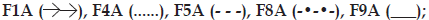 (b) Effect of excipients on in vitro gel
erosion from transdermal formulations:
(b) Effect of excipients on in vitro gel
erosion from transdermal formulations: 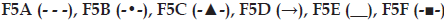 .
.
F1A, F5A and F9A release profiles (fig. 1a) were compared for the effect of polymer concentration, and the release profile of F1A was found similar with F5A, although the mechanical properties of these gel bases (F1, F5, F9) were significantly different (Table 2). Mechanical properties and gelation temperatures of F9, which has the lowest F127 concentration, was significantly lower than F1 and F5 bases and this was also reflected to in vitro release (fig. 1a) as a significantly higher release. Decrease in polymer concentration facilitate the application of gel from a container, however, it affects release profiles negatively especially for F9. This dramatic change was also observed in erosion graphics given for F1A, F5A and F9A (fig. 2a). F9A prepared with the lowest polymer concentration gave high erosion, as expected.
Effect of different polymers (S-97, K100LV), solubility enhancers (DMSO, PEG 400) and salt (NaCl) on in vitro drug release and gel erosion is given in figs. 1b and 2b. According to these graphics, only K100LV (F5C) decreased the meloxicam release of from F5A. The release profile of F5C can be explained as; initial water absorption of K100LV leading a swelled, strengthen and saturated layer at gel surface, which extended the diffusion way until 120 min and retarded the total erosion of the F5C and decelerated the release of drug (figs. 1b and 2b). These findings are in accordance with the study of Cavallari et al., which presented the relation between the high ratio of HPMC and slower release of drug due to gel barrier forming upper surface of formulation [22]. Additionally dehydration mechanism, which caused aggregation of PPO units, resulted as a micellization in poloxamer solutions in the existence of hydrophilic HPMC and it was assumed that the interaction between the network of poloxamer micelles and HPMC resulted as a high resistance in situ gel that retarded the erosion [23,24]. As a result, a complicated mechanism including longer diffusion way due to swelling and erosion-dependent release due to the poor water solubility of drug caused a slower release profile of drug compared with other formulations.
Evaluation of figures together with texture data (figs. 1b, 2b and Table 2) showed that the existence of PEG 400 (F5E) did not significantly change the release profile of the drug, but lowered the erosion of the gel compared with F5A. This could be attributed to the stabilising effect of PEG 400 that straighten the texture properties by increasing viscosity, hardness, compressibility and adhesiveness but lowering CMT of the formulation (Table 2). Furthermore PEG 400 is known as a good solubilising agent for meloxicam. According to Cavallari et al. [22], as a plasticiser, PEG 400 had the property easier distension of polymer chains, which causes to be quick release of drug owing to faster diffusion through the formulation, thus leading to higher rates of drug release. Water soluble PEG 400 also operates a partial erosion of the formulation due to dissolution of itself [22].
Although NaCl is used for increasing viscosity, which leads to strengthen gel [13] and expected to cause slower erosion and release of drug, it caused no decrease in release of drug and showed an increase after 180 min (fig. 1b). This could be attributed to the pore forming effect of NaCl, which caused a sudden increase in the gel erosion profile of F5F probably due to disintegration of water filled channels in the gel structure after 300 min (fig. 2b). Although NaCl has reinforced gel strength and bioadhesive force especially over 1% w/w concentration [13], it has decreased the CMT due to its salting in effect related the dehydration of PPO blocks [22] presented in some studies [25,26]. The addition of 1% w/w NaCl decreased approximately 3° of CMT as presented in Table 2.
F5B gave the highest release of drug due to the presence of S-97, which hindered gel formation in the temperature range studied (fig. 2b). Although previous studies indicate that viscous solutions of S-97 at pH 6.8 caused ionisation of carboxylic acid groups, thus retards the release of drugs [21] and S-97 swelled at pH 7.4 [27], in our study, we obtain a fast release with S-97, which could be attributed to the nongelation of the formulation, thus F5B gave a viscous but weak gel under body temperature (Table 3). Erosion was the highest for F5B compared with others, which was in accordance with highest release (figs. 1b and 2b).
| Code | F1A | F4A | F5A | F8A | F9A | F5B | F5C | F5D | F5E | |
|---|---|---|---|---|---|---|---|---|---|---|
| r2 | (linear) | 0.814 | 0.999 | 0.943 | 0.917 | 0.991 | 0.999 | 0.864 | 0.933 | 0.678 |
| r2 | (polynomial) | 0.998 | 0.999 | 0.961 | 0.952 | 0.999 | 0.999 | 0.875 | 0.949 | 0.68 |
Table 3: Data Analysis Of Gel Erosion Versus Drug Release From The Gel Formulations
Existence of topical enhancer DMSO in F5D resulted as reduce in the intensity of micellization (Table 2), probably due to the DMSOs ability to disrupt water structures and thus modulate the water shells around the PPO groups of F127. A linear release profile of hydrophobic drug obtained for 6 hours with F5D (fig. 1b), which was supported by the study of Ur-Rehman et al. [14]. Although DMSO increased the gel strength, thus hindered the erosion of F5D (fig. 2b), it also increased release of hydrophobic drug (fig. 1b) probably due to the solubility enhancing effect [28].
Data analysis for release kinetics showed that (Table 4), all of the formulations generally fitted to both Zero-order and Korsmeyer–Peppas kinetics, according to their high r2 values. High r2 values were obtained for Higuchi model as well. Moore et al. indicated that, drug release and gel dissolution from poloxamer gels generally fitted to Zero-order, at least for the first 90% of the release process [29]. High r2 values of Korsmeyer–Peppas kinetics obtained for formulations can be used to describe the drug release and erosion relationship of formulations. According to obtained diffusional exponent values of formulations, while F4A and F5C indicated non-Fickian (0.45<n<0.89) diffusion mechanism of drug release, which was supported by their gel erosion profile, all the other formulations indicated Case II transport (0.89<n<1.0) in another expression zero order mechanism, which was supported by their release and gel erosion profiles (figs.1b and 2b) [30,31]. Zero-order rate constants (k) of F1A, F5A and F9A formulations were not significantly different, while F4A and F8A showed significant difference that can be attributed to the presence of F68 polymer in formulations, which significantly changed k values compared with F1A and F5A formulations. These were in accordance with the findings in fig. 1. The highest value of k was obtained with F5B including S-97 polymer and the slowest value of k, accordingly release of drug, achieved with F5C owing to HPMC polymer swelling property.
| Code | First | Zero | Higuchi | Korsmeyer– | ||||
|---|---|---|---|---|---|---|---|---|
| order | order | Peppas | ||||||
| r2 | k | r2 | k | r2 | k | r2 | n | |
| F1A | 0.9308 | 0.0044 | 0.988 | 0.2337 | 0.99 | 6.738 | 0.9921 | 1.001 |
| F4A | 0.9283 | 0.0067 | 0.9849 | 0.264 | 0.9708 | 6.987 | 0.8329 | 0.8307 |
| F5A | 0.9902 | 0.0045 | 0.9937 | 0.2249 | 0.9519 | 6.004 | 0.9914 | 0.9386 |
| F8A | 0.9994 | 0.0086 | 0.9567 | 0.3776 | 0.9618 | 7.926 | 0.9999 | 1.093 |
| F9A | 0.9658 | 0.0045 | 0.9778 | 0.2291 | 0.9763 | 5.987 | 0.9963 | 0.9758 |
| F5B | 0.9142 | 0.0125 | 0.9996 | 0.5034 | 0.9836 | 9.407 | 0.9991 | 1.015 |
| F5C | 0.9712 | 0.003 | 0.9894 | 0.18 | 0.9637 | 4.582 | 0.9956 | 0.8487 |
| F5D | 0.9141 | 0.0588 | 0.9989 | 0.2764 | 0.9764 | 7.304 | 0.9973 | 0.8921 |
| F5E | 0.9065 | 0.0042 | 0.9925 | 0.2209 | 0.9554 | 6.058 | 0.9908 | 1.025 |
| F5F | 0.8314 | 0.006 | 0.9888 | 0.2544 | 0.9601 | 7.175 | 0.9983 | 1.212 |
Table 4: Release Kinetics Of Meloxicam From Gel Formulations
Regression data obtained from gel erosion versus drug release graphics used to show the relation between erosion and drug release (Table 3). This relation was both investigated for linear and polynomial due to the nonlinear erosion data of highly swellable formulations (such as high polymer concentration or the existence of HPMC). According highest r2 values obtained, erosion was found to be effective on drug release from F4A including F68; F9A having low polymer concentration and F5B including S-97, which was more rapidly eroded thus gave a high r2 value. Higher polymer concentration (F1A) or the existence of additives increasing gel strength (F5C, F5E, F5F) had relatively low r2 values. Low r2 value of F5C should also be attributed to high swelling property of K100LV polymer used in this formulation.
In conclusion, a promising transdermal poloxamer based in situ gelling formulation could be developed for joint disease. Generally, it is expected to prepare a gel soft enough to get from the container, but the firmness of the gel affects the release of drugs and in case of poor firmness release would be faster, according to erosion mechanism involved with poloxamers. This study indicated that a sustained release of meloxicam for 6 h of which is the usual minimum limit for sleeping from a quite hard gel formulation could be achieved with F127-K100LV based gel formulation. Although F127-K100LV combination tends to precipitate in weak gels with low gel strength, the polymer concentration in this study did not give a solution under body temperature and the formulations prepared was stored as a homogeneous gel in room temperature; thus, precipitation was not a problem in our study. F127–K100LV combination with low dose meloxicam could be improved by further studies as a transdermal formulation for applying during night time, thus enhance the bioavailability of drugs due to circadian rhythm for rheumatoid arthritis and their chronotherapy.
Acknowledgements
The authors would like to thank Prof. Dr. Tamer Baykara for his kind material support. This study is part of a project supported by Ankara University Scientific Project Office-BAP (10B3336004).
References
- Shin SC, Cho CW, Oh IJ. Enhanced efficacy by percutaneous absorption of piroxicam from the poloxamer gel in rats. Int J Pharm 2000;193:213-8.
- Seti P, Kruss B, Wiegleb J, Trach V. Local tissue tolerability of meloxicam, a new NSAID: Indications for parental, dermal and mucosal administration. Br J Rheumatol 1996;35:44-50.
- Ah YC, Choi JK, Choi YK, Ki HM, Bae JH. A novel transdermal patch incorporating meloxicam: In vitro and in vivo characterization. Int J Pharm 2010;385:12-9.
- Bastien M, Milpied-Homsi B, Baudot S, Dutartre H, Litoux P. Ketopofen induced contact photosensitivity disorders; 5 cases. Ann Dermatol Venereol 1997;124:523-6.
- Bruguerolle B, Labrecque G. Rhythmic pattern in pain and chronotherapy. Adv Drug Del Rev 2007;59:883-95.
- Liaw J, Lin YC. Evaluation of poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) (PEO–PPO–PEO) gels as a release vehicle for percutaneous fentanyl. J Control Rel 2000;68:273-82.
- Algın Yapar E, İnal Ö. Poly(ethylene oxide)–poly(propylene oxide) based copolymers in transdermal drug delivery: An overview. Tropical J Pharm Res 2012;11:855-66.
- Chiappetta DA, Sosnik A. Poly(ethylene oxide)–poly(propylene oxide) block copolymer micelles as drug delivery agents: Improved hydrosolubility, stability and bioavailability of drugs. Eur J Pharm Biopharm 2007;66:303-17.
- Ruel-Garie´py E, Leroux JC. In situ-forming hydrogels: Review of temperature-sensitive systems. Eur J Pharm Biopharm 2004;58:409-26.
- Pandit NK, Wang D. Salt effects on the diffusion and release rate of propranolol from poloxamer 407 gels. Int J Pharm 1998;167:183-9.
- Dumortier G, Groissord JL, Agnely F, Chaumeil JC. A review of poloxamer 407 pharmaceutical and pharmacological characteristics. Pharm Res 2006;23:2709-28.
- Xiong XY, Tam KC, Gan LH. Polymeric nanostructures for drug delivery applications based on Pluronic copolymer systems. J Nanosci Nanotechnol 2006;6:2638-50.
- Yong SY, Choi SJ, Quan QZ, Rhee JD, Kim CK, Lim SJ, et al. Effect of sodium chloride on gelation temperature, gel strength and bioadhesive force of poloxamer gels containing diclofenac sodium. Int J Pharm 2001;226:195-205.
- Ur-Rehman T, Tavelin S, Gröbner G. Effect of DMSO on micellization, gelation and drug release profile of poloxamer 407. Int J Pharm 2010;394:92-8.
- Schmolka IR. Artificial skin. I. Preparation and properties of pluronic F-127 gels for treatment of burns. J Biomed Mater Res 1972;6:571-82.
- Jones DS, Woolfson AD, Brown AF. Textural analysis and flow rheometry of novel, bioadhesive antimicrobial oral gels. Pharm Res 1997;14:450-7.
- Amasya G, Karavana SY, Şen T, Baloğlu E, Tarımcı N. Bioadhesive and mechanical properties of triamcinolone acetonide buccal gels. Turk J Pharm Sci 2012;9:1-12.
- Zhang L, Parsons DL, Navarre C, Kompella UB. Development and in vitro evaluation of sustained release Poloxamer 407 (P407) gelformulations of ceftiofur. J Control Rel 2002;85:73-81.
- Baloğlu E, Yaprak-Karavana S, Ay-Senyigit Z, Guneri T. Rheological and mechanical properties of poloxamer mixtures as a mucoadhesive gel base. Pharm Dev Tech 2011;16:627-36.
- Asasuratjit R, Thanasandrokpibull S, Fuangfuchat A, Veeranondha S. Optimization and evaluation of thermoresponsive diclofenac Na opthalmic in situ gels. Int J Pharm 2012;411:128-35.
- Dhiman M, Yedurkar P, Sawant KK. Formulation, characterization and in vitro evaluation of bioadhesive gels containing 5-Fluorouracil. PharmDev Tech 2008;13:15-25.
- Cavallari C, Fini A, Ospitali F. Mucoadhesive multiparticulate patch for the intrabuccal controlled delivery of lidocaine. Eur J Pharm Biopharm 2013;83:405-14.
- Koffi AA, Agnely F, Ponchel G, Grossiord JL. Modulation of the rheological and mucoadhesive properties of thermosensitive poloxamer-based hydrogels intended for the rectal administration of quinine. Eur J Pharm Sci 2006;27:328-35.
- Conti S, Maggi L, Segale L, Ochoa Machiste E, Conte U, Grenier P, et al. Matrices containing NaCMC and HPMC 2. Swelling and releasemechanism study. Int J Pharm 2007;333:143-51.
- Pandit N, Trygstad T, Croy S, Bohorquez M, Koch C. Effect of salts on the micellization, clouding, and solubilization behaviour of Pluronic 127 solutions. J Colloid Interface Sci 2000;222:213-20.
- Yuan Y, Ying C, Li Z, Hui-Ping Z, Yi-Sha G, Bo Z,et al. Thermosensitive and mucoadhesive in situ gel based on poloxamer as new carrier for rectal administration of nimesulide. Int J Pharm 2012;430:114-9.
- Plochocka, Krystyna, Lynn, Jeffrey A. Polymeric Hydrogels. United States Patent, No: US, 6,583,225 B1, 2003.
- Jantharaprapap R, Stagni G. Effect of penetration enhancers on in vitro permeability of meloxicam gels. Int J Pharm 2006;343:26-32.
- Moore T, Croy S, Mallapragada S, Pandit N. Experimental investigation and mathematical modeling of Pluronic F127 gel dissolution: Drug release in stirred systems. J Control Release 2000;67:191-202.
- Conti S, Maggia L, Segalea L, Ochoa Machiste E, Conte U, Grenier P, et al. Matrices containing NaCMC and HPMC 1. Dissolution performance characterization. Int J Pharm 2007;333:136-42.
- Nasir F, Iqbal Z, Khan JA, Khan A, Khuda F, Ahmad L, et al. Development and evaluation of diclofenac sodium thermoreversible subcutaneous drug delivery system. Int J Pharm 2012;439:120-6.





