- *Corresponding Author:
- S. K. Kashaw
Department of Pharmaceutical Sciences, Dr. Harisingh Gour University (A Central University), Sagar-470 003, India
E-mail: sushilkashaw@gmail.com
| Date of Submission | 22 November 2018 |
| Date of Revision | 17 January 2019 |
| Date of Acceptance | 31 May 2019 |
| Indian J Pharm Sci 2019;81(4):790-796 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
Benzylamine derivatives have recently been demonstrated interesting potential as the treatment for chronic obstructive pulmonary disease via interaction with the PDE4 enzymes. To understand the necessity around the nucleus k-nearest-neighbour molecular field analysis-based 3D-QSAR analysis was performed on a series of 47 compounds. The 3D-QSAR studies were performed using stepwise variable selection k-nearest-neighbour molecular field analysis approach; a leave-one-out cross-validated correlation coefficient (q2) of 0.8805 and a predicted r2 for the external test (pred_r2) of 0.6677 were obtained. Points generated in k-nearest-neighbour molecular field analysis 3D-QSAR model were S_1789 (-0.0023, -0.0021), E_14 (0.0271, 0.03699). Results showed positive range indicating that positive electrostatic potential is favourable for increase in the activity and hence a less electronegative substituent group is preferred in lattice point 14 (around R1). Negative range indicates that negative steric potential is favourable for increase in the activity and hence less bulky substituent group is preferred at lattice point. The information rendered by 3D-QSAR models lead to a better understanding of structural requirements for PDE4 inhibitors and help in the design of novel potent molecules.
Keywords
kNN-MF 3D-QSAR, PDE4 inhibitors, COPD
Chronic obstructive pulmonary disease (COPD) is a syndrome that includes chronic bronchitis and emphysema. Currently it is one of the leading causes of morbidity and mortality around the world. It is a major public health burden, with a global prevalence of 10.1 %, causing 3 million deaths worldwide in 2011 and predicted to be the fourth leading cause of death by 2030[1]. The financial burden due to this disease is around $ 49.8 billion in 2010 in the US[2]. Presently long-acting bronchodilators (LABA), long-acting muscarinic antogonists (LAMA), and NSAIDs are employed to counteract the manifestations of COPD[3-6]. The phosphodiesterase (PDE) family, represented by four genes, PDE4A, -B, -C, and -D, causes the hydrolysis of phosphodiester bond of cAMP to yield inactive AMP. These in turn reduce a wide variety of inflammatory mediators and inflammatory cell functions. PDE4 inhibitors reduce pulmonary inflammation and improve lung function in preclinical models of asthma and COPD[7-11]. Roflumilast and cilomilast are some of the PDE4 inhibitors under development, however, their use is limited due to dose-dependent toxicities[12]. Majority of research lacks a high-resolution structure of the target protein i.e. membrane spanning G-proteincoupled receptors and membrane transporters. Nine out of twenty most selling prescribed molecules in the world belongs to these categories of compounds in year 2000[13]. For these cases, significant predictive models of ligand binding to target active sites can be useful to analyse the correlation between biological activity and physicochemical properties.
3D-QSAR is broadly used tool to recognize the steric, electrostatic, and hydrophobic interactions of various drugs acting via receptor modulation for exerting biological activity[14,15]. The focus of the present work was to develop the 3D-QSAR-based k-nearestneighbour molecular field analysis (kNN-MF) studies on benzylamine derivatives. The model was validated for their regression coefficient, internal and external predictive ability and statistical significance. Further, developed model was interpreted to investigate the contribution of various 3D descriptors as PDE4 inhibitors.
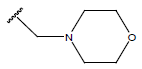
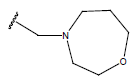
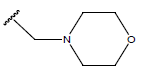
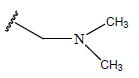
All computational studies were performed on Microsoft Windows 7 running on Pentium processor by using VlifeMDS (version 4.4) molecular modeling software[16]. In the present study a set of 47 molecules of the class of benzylamine derivatives were synthesised and evaluated by Savi et. al[17]. The compounds of the series were reported to be effective against COPD. The biological evaluation was performed on human PDE4B enzyme and the results were expressed in terms of pIC50 value represented in Table 1.
 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | pIC50 value | Compound | R1 | R2 | R3 | pIC50 value | ||
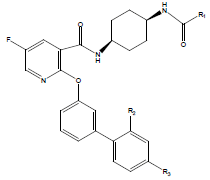
| 01 | 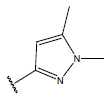 |
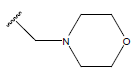 |
-OH | 10.8 | 25. | 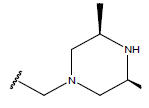 |
H | 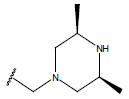 |
10.6 | ||
| 02 | 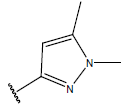 |
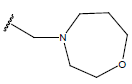 |
-OH | 11 | 26. | 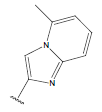 |
H | 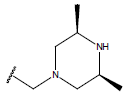 |
10.2 | ||
| 03 | 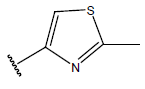 |
 |
-OH | 10.6 | 27. | 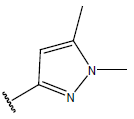 |
H | 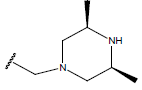 |
10.6 | ||
| 04 | 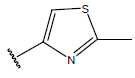 |
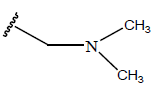 |
-OH | 10.5 | 28. | 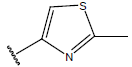 |
H | 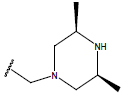 |
10.2 | ||
| 05 | 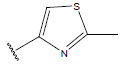 |
 |
-OH | 10.7 | 29. | 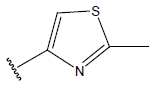 |
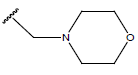
H | 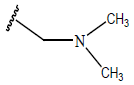 |
9.4 | ||
| 06 | 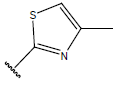 |
 |
-OH | 10.8 | 30. | 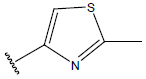 |
H | 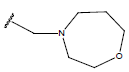 |
10 | ||
| 07 | 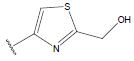 |
 |
-OH | 10.2 | 31. | 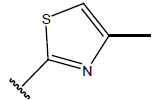 |
H | 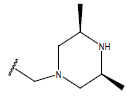 |
10.5 | ||
| 08 | 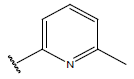 |
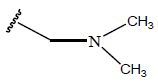 |
-OH | 10.5 | 32 | 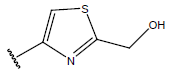 |
H | 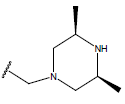 |
10.1 | ||
| 09 | 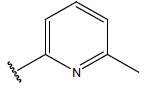 |
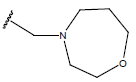 |
-OH | 10.7 | 33 | 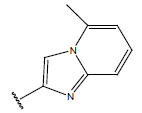 |
H | 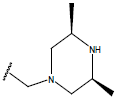 |
10.2 | ||
| 10 | 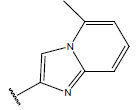 |
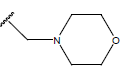 |
-OH | 10.8 | 34 | 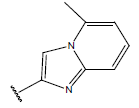 |
H | 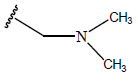 |
9.1 | ||
| 11. | 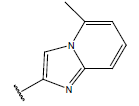 |
 |
-OH | 10.3 | 35 | 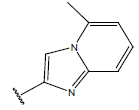 |
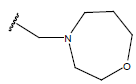
H | 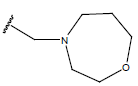 |
9.9 | ||
| 12. | 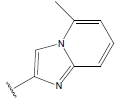 |
 |
-OH | 10.5 | 36 | 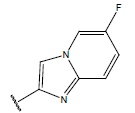 |
H | 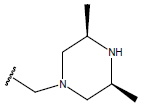 |
10 | ||
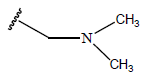
| 13. | 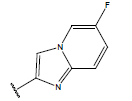 |
 |
-OH | 10.3 | 37 | 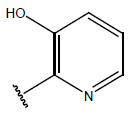 |
H | 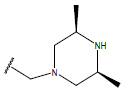 |
9.9 | ||
| 14 | 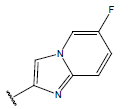 |
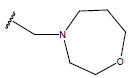 |
-OH | 10.7 | 38 | 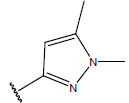 |
H | 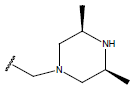 |
10.6 | ||
| 15 | 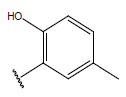 |
 |
-OH | 10.6 | 39 | 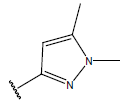 |
-OH | 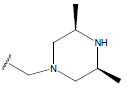 |
10.5 | ||
| 16. | 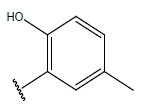 |
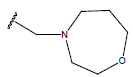 |
-OH | 10.5 | 40 | 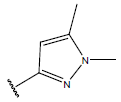 |
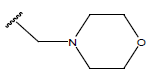
H | -OH | 10.2 | ||
| 17. | 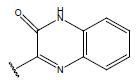 |
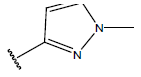 |
-OH | 9.5 | 41 | 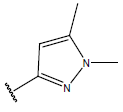 |
H | 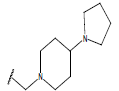 |
10.7 | ||
| 18. | 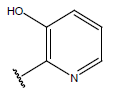 |
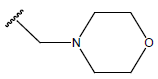 |
-OH | 9.9 | 42 | 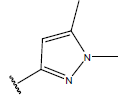 |
H | 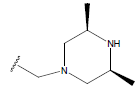 |
10.5 | ||
| 19. | 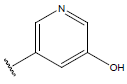 |
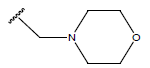 |
-OH | 8.6 | 43 | 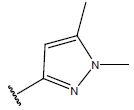 |
-OH | H | 10.1 | ||
| 20. | 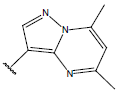 |
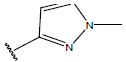 |
-OH | 10.3 | 44 | 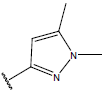 |
H | 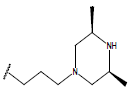 |
10.7 | ||
| 21. | 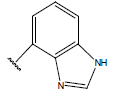 |
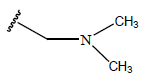 |
-OH | 10.3 | 45 | 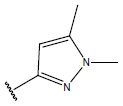 |
H | 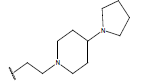 |
10.4 | ||
| 22. | 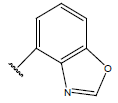 |
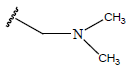 |
-OH | 9.5 | 46 | 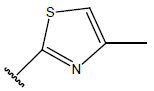 |
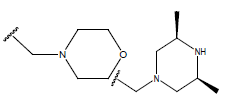 |
10.6 | |||
| 23. | 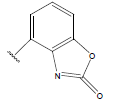 |
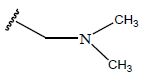 |
-OH | 7.7 | 47 | 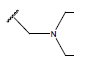 |
 |
10.7 | |||
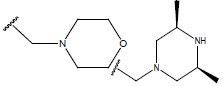
| 24. | 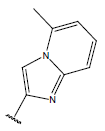 |
H |  |
10.4 | |||||||
Table 1: Structure of the compounds f and their biological activity data
The set of molecules considered in this study were sketched using the 2D draw application and converted to 3D structures. The energy of all the structures were optimised by Merck Molecular Force Field (MMFF) method with 10 000 00 as maximum number of cycles, 0.001 cal/molÅ as convergence criteria and dielectric constant was fixed at 1 as constant. The cut-off for electrostatic and steric energy was set to 30.0 and 10.0 kcal/mol. Energy minimization was carried out by a molecular mechanics programme, which calculates the energy of the starting molecules then varies the bond lengths, bond angles and torsion angles to create new structure having stable state.
In 3D QSAR, molecular alignment was done to develop counter plot. For accurate alignment, conformers were generated by Monte Carlo conformational search method and the conformers of least energy were selected. All the compounds were aligned by template based method, (where, a template was built by considering common substructures in the series) using most active compound 23 as reference molecule. In present study, all the compounds were aligned against minimum energy conformation of most active compound 23 by using 5-fluoro-nicotinamide moiety as template shown in fig. 1A. The aligned molecules (fig. 1B) were selected for calculation of descriptors by inserting biological activity as dependent variable.

Figure 1: Molecular alignment of benzylamine derivatives
A. Template for molecular alignment and B. template-based alignment of benzylamine derivatives
Once the molecules are aligned, the QSAR model was built by kNN-MFA method using forward-backward as variable selection method. Various training and test set of benzylamine derivatives were constructed using manual data selection. Compounds were divided in training and test set in a way that biological activities of all compounds in test set lie within the maximum and minimum value range of biological activities of training set of compounds. Fitness plot for model shown in fig. 2 where, training set (red points) as well as test set (blue points) were well closed to regression line and showing good predictive ability of the model. Fig. 3 indicate the radar plot of training and test set, which showed closeness between actual and predicted biological activity of compounds.

Figure 2: Fitness plot of observed vs. predicted activity
( ) Training, (
) Training, ( ) test
) test

Figure 3: Radar plot of training and test set
A: Test; B: training, () actual, () predicted
In 3D-QSAR analysis steric, electrostatic and hydrophobic fields were computed at lattice points of the grid using a methyl probe of charge +1. This develops rectangular grid that optimize the molecular fields required around the compounds to act on the PDE4 receptors.
3D QSAR models were generated using k-NN-MFA coupled with stepwise forward-backward variable selection method. Values of selected 3D-QSAR model were statistically significant and showed good predictive power. The best model generated with kNN-MFA method showed a q2, pred_r2 and K nearest neighbour as 0.8805, 0.6677 and 2, respectively as shown in Table 2. The contour plot of the kNN-MFA (fig. 4) shows the relative position and ranges of the corresponding important electrostatic/steric fields in the model. Points generated in kNN-MFA 3D-QSAR model were S_1789 (-0.0023, -0.0021), E_14 (0.0271, 0.03699), i.e. steric and electrostatic data points at lattice points 1789 and 14, respectively. The ranges of data point values were based on the variation of the field values at the chosen points using the most active molecule and its nearest neighbour set. Positive range indicates that positive electrostatic potential is favourable for the enhancement of the biological activity and hence a less electronegative substituent group is preferred at lattice point 14 (i.e. around R1). Negative range indicates that negative steric potential is favourable for increase in the activity and hence less bulky substituent group is preferred at lattice point 1789 (i.e.R2 and R3). Fig. 5 describes outline for the development of new benzylamine derivatives to fabricate better and effective drugs for COPD treatment.
| MODEL SUMMARY | |
|---|---|
| kNN Method | |
| Training Set Size = 32 | |
| Test Set Size = 15 (01, 02, 06, 08, 12, 13, 15, 17, 21, 30, 33, 44, 45, 46) | |
| Statistics: | |
| k Nearest Neighbour | 2 |
| n | 32 |
| Degree_of_freedom | 29 |
| q2 | 0.8805 |
| q2_se | 0.3823 |
| pred_r2 | 0.6677 |
| pred_r2se | 0.2489 |
| Descriptor Range: | |
| E_14 (0.0271, 0.03699) | |
| S_1789 (-0.0023, -0.0021) | |
Table 2: Statistical results of 3D QSAR model

Figure 4: Relative positions of the local fields around aligned molecules

Figure 5: Diagrammatic representation of the required pharmacophoric pattern around benzylamine derivative for optimum activity
In conclusion, the model developed to predict the structural features around benzylamine derivatives for COPD treatment reveals useful information for designing new molecules. The contour plot obtained for the 3D model indicates that positive contribution of electrostatic potential and negative contribution of steric potential is required for improved potency of benzylamine derivatives for COPD treatment. Thus, on the basis of molecular fields generated in 3D model could be employed to design new derivatives with more potent inhibitory activity.
Acknowledgement:
One of the authors Priya Singh wishes to thank the AICTE for providing financial support to undertake this project.
Conflict of interest:
Authors declare no conflicts of interest.
References
- Mathers CD, Loncar D. Projections of Global Mortality and Burden of Disease from 2002 to 2030. PLoS Med 2006;28:209-24.
- Adeloye D, Chua S, Lee C, Basquill C, Papana A, Theodoratou E, IJPS-17-353 (571). Global and regional estimates of COPD prevalence: Systematic review and meta-analysis. J Glob Health. 2015;5(2):020415.
- Sundar IK, Nevid MZ, Friedman AE, Rahman I. Cigarette smoke induces distinct histone modifications in lung cells: implications for the pathogenesis of COPD and lung cancer. J Proteome Res 2014;13:982-96.
- Press NJ, Taylor RJ, Fullerton JD, Tranter P, McCarthy C, Keller TH, et al. Discovery and Optimization of 4-(8-(3-Fluorophenyl)-1,7-naphthyridin-6-yl)transcyclohexane carboxylic acid, an improved PDE4 inhibitor for the treatment of Chronic Obstructive Pulmonary Disease (COPD). J Med Chem 2015;58:6747-52.
- Danielle C, Buys N, Sriram KB, Sharma S, Morris N, Sun J. The effects of chronic obstructive pulmonary disease self-management interventions on improvement of quality of life in COPD patients: A meta-analysis. Respir Med 2016;121:81-90.
- Kim I, Song JH, Park CM, Jeong JW, Kim HR, Ha JR, et al. Design, synthesis and evaluation of 2-aryl-7-(3’,4’-dialkoxyphenyl)-pyrazolo [1,5-a] pyrimidines as novel PDE-4 inhibitors. Bioorg Med Chem Lett 2010;20:922-26.
- Mey MV, Bommelé KM, Boss H, Hatzelmann A, Van SM, Timmerman SH. Synthesis and structure-activity relationships of cis-tetrahydrophthalazinone/pyridazinone hybrids: A novel series of potent dual PDE3/PDE4 inhibitory agents. J Med Chem 2003;46:2008-16.
- Luan VT. Fourth-Order Two-Stage Explicit Exponential Integrators For Time-Dependent Pdes. Appl Numer Math 2017;112:91-103.
- Balasubramanian G, Narayanan S, Andiappan L, Sappanimuthu T, Thirunavukkarasu S, Sundaram S, et al. In vivo Effective Dibenzo[B,D]Furan-1-Yl-Thiazoles As Novel Pde-4 Inhibitors. Bioorg Med Chem 2016;24(22):5702-16.
- Calzetta L, Cazzola M, Page CP, Rogliani P, Facciolo F, Matera MG. Pharmacological characterization of the interaction between the dual phosphodiesterase (PDE) 3/4 inhibitor RPL554 and glycopyrronium on human isolated bronchi and small airways. Pulm Pharmacol Ther 2015;32:15-23.
- Praveena KSS, Durgadas S, Babu NS, Akkenapally S, Kumar CG, Deora GS, et al. Synthesis of 2,2,4-trimethyl-1,2-dihydroquinolinyl substituted 1,2,3-triazole derivatives: their evaluation as potential PDE 4B inhibitors possessing cytotoxic properties against cancer cells. Bioorg Chem 2014;53:8-14.
- Rabe KF. Update on roflumilast, a phosphodiesterase 4 inhibitor for the treatment of chronic obstructive pulmonary disease. Br J Pharmacol 2011;163(1):53-67.
- Ahluwalia VK, Chopra M. Medicinal Chemistry. 1st ed. Boca Raton, Florida, United States: CRC Press; 2008. p. 1-21.
- Bhatiya R, Vaidya A, Kashaw SK, Jain AK, Agrawal RK. QSAR analysis of furanone derivatives as potential COX-2 inhibitors: kNN MFA approach. J Saudi Chem Soc 2014;18:977-84.
- Mishra M, Mishra VK, Senger P, Pathak AK, Kashaw SK. Exploring QSAR studies on 4-substituted quinazoline derivatives as antimalarial compounds for the development of predictive models. Med Chem Res 2014;23:1397-405.
- Molecular Design Suite (2004), V-life Sciences Technologies Pvt. Ltd, Pune, India. Available from: https://www.vlifesciences.com/products/QSARPro/Product_QSARpro.php.
- De Savi C, Cox RJ, Warner DJ, Cook AR, Dickinson MR, McDonough A, et al. Efficacious inhaled PDE4 inhibitors with low emetic potential and long duration of action for the treatment of COPD. J Med Chem 2014;57:4661-76.





