- *Corresponding Author:
- B. Mohammadi
Medical Biology Research Center, Kermanshah University of Medical Sciences, Kermanshah, Iran
E-mail: baharbmk2010@yahoo.com
| Date of Submission | 08 August 2011 |
| Date of Revision | 10 August 2012 |
| Date of Acceptance | 16 August 2012 |
| Indian J Pharm Sci, 2012, 74 (4): 360-364 |
Abstract
An accurate and very rapid method for determination of zonisamide an antiepileptic drug, in human serum is described. The analytical procedure involves liquid-liquid extraction of the analyte and an internal standard (vanillin) from human serum by ethyl acetate as extracting solvent. Chromatographic separation was achieved using a monolithic C18 analytical column and a mixture of 0.05 M phosphate buffer containing triethylamine (1 ml/l; pH 2.7) and methanol (83:17 v/v) was used as the mobile phase. The detection wavelength was set at 240 nm. The calibration curve was linear over a concentration range of 0.015-6.4 μg/ml of zonisamide in human serum. The total run time of analysis was 3.5 min and the lower limits of detection and quantification were 0.005 and 0.015 μg/ml, respectively. The method validation was carried out in terms of specificity, sensitivity, linearity, precision, accuracy and stability. The validated method was applied in a randomised crossover bioequivalence study of two different zonisamide preparations in 24 healthy volunteers, and the assay was sensitive enough to measure drug levels up to 8 days following a single dose administration of zonisamide.
Keywords
Bioequivalence study, chromatography, high performance liquid chromatography, zonisamide
Zonisamide (1,2?benzisoxazole?3?methanesulfonamide) is a new structurally unique antiepileptic drug (AED), which provides highly effective adjunctive treatment of refractory partial seizures and is also used to treat neurological diseases such as Parkinson, bipolar disorder and migraine headaches [1?5]. The drug acts by blockade of voltage?sensitive sodium and voltage?gated calcium channels [6]. Zonisamide has a good bioavailability (95%) with therapeutic concentrations of 10?30 μg/ml, linear kinetic and long terminal half life (approximately 63 h) [7,8]. The long half life of zonisamide reduces the need for routine monitoring of blood levels in most patients [9]. In human single dose pharmacokinetic studies, however, a sensitive method is needed to follow plasma concentration?time profiles up to three half lives postdose (about 8 days). Several analytical methods for analysis of zonisamide including liquid chromatography (LC)?mass [10] voltametric method [11], gas chromatography (GC) [12], micellar electrokinetic capillary chromatography [13], enzyme immunoassay [14], high performance liquid chromatography (HPLC) have been published to quantify zonisamide either alone [15?18], or simultaneous with other AEDs [19?22]. However, these methods neither have been tested in the single?dose bioequivalence studies, nor are sensitive enough to measure the drug concentrations up to three half?lives after a single dose administration. The present article describes simple, very fast and sensitive method for analysis of zonisamide in human blood samples using HPLC with ultraviolet detection. This method was validated and applied in a randomised crossover bioequivalence study in 24 healthy volunteers is sufficiently sensitive to follow plasma concentration?time profiles of the drug up to 180 h postdose following single dose administration.
Zonisamide (purity 99.6%) was from Dipharma, Italy. Vanillin (IS) was purchased from Merck, Darmstand, Germany. All reagents used were of analytical grade except methanol which was HPLC grade. Water was glass?double distilled, and further purified for HPLC with a Maxima purification system (USF ELGA, England). The HPLC system used comprised two pumps of Shimadzu LC?10A solvent delivery system, a system controller (SCL 10AD), a UV/Vis spectrophotometric detector (SPD?10AD) operated at wavelength of 240 nm, a column oven (CTO?10A) and a data processor (C?R4A) all from Shimadzu, Kyoto, Japan.
A stock solution of zonisamide (1000 μg/ml) was prepared in methanol. Working standards of the drug (0.15?64 μg/ml) were prepared by serial dilution of the stock solution in methanol. Working standard solution of the IS (50 μg/ml) was prepared in methanol. All solutions were stored at 4° and were stable for at least 3 weeks.
A mixture of 0.05 M sodium phosphate buffer containing triethylamine (1 ml/l; pH 2.7; adjusted with phosphoric acid) and methanol (83:17 v/v) was used as the mobile phase and analytical separation was achieved on a Chromolith performance RP 18 monolithic HPLC column (100×3 mm ID). The column oven temperature was set at 35° and the mobile phase was filtered, degassed and pumped at a flow rate of 3.2 ml/min.
Serum samples (1 ml, unknown, blank or calibration) were transferred into 100×16 mm disposable glass tubes. To each of these tubes, 100 μl of the I.S. (50 μg/ml) was added and after a brief mixing, the analytes were extracted with 4 ml ethyl acetate. After vortex mixing for 30 s and centrifugation (5 min at 6000 ×g), the organic phase was removed and evaporated to dryness under stream of nitrogen at 50°. The residue was reconstituted in 100 μl mobile phase and following brief mixing a volume of 20 μl was injected onto the HPLC.
Calibration curves (n=5) samples were prepared within the concentration range of 0.015?6.4 μg/ml using pooled human blank serum obtained from healthy subjects. In disposable glass tubes (100×16 mm), after evaporation of 100 μl from each working solutions of the analyte, under a gentle stream of nitrogen at 50°, the residues were reconstituted in 1 ml of drug?free human serum, mixed for 10 s on a vortex mixer and subjected to extraction and analysis as described above. The linearity of the method was checked in the same day (n=6) and in 10 consecutive days. Calibration curves (weighted regression line) were obtained by linear least?squares regression analysis of plots of peak?area ratio to I.S. versus drug concentrations. Quality control samples used in method validation were prepared with the drug working solutions to make low (0.015 μg/ml), medium (1.5 μg/ml) and high (6.4 μg/ml) concentrations of the analyte. Intra? and inter?day variations were calculated by repeated analysis (n=6) of different concentrations of zonisamide in a single analytical run and in 10 analytical runs performed on different days, respectively. Limit of detection (LOD) and limit of quantification (LOQ) were defined as the concentrations of the drug giving a signal?to?noise ratio of 3:1 and 10:1, respectively. The specificity of the method was examined by the presence of disturbing endogenous peaks in 24 human serum samples from different volunteers. Samples extraction were achieved according to the sample preparation procedure, except for the addition of the I.S. The extraction efficiencies of zonisamide at the above mentioned concentrations as well as the I.S. at applied concentration were calculated in replicates (n=5) by comparing the respective peak areas obtained by extraction of the samples from serum, with those obtained from the same amounts of unextracted solutions in methanol. Selectivity of the assay was examined by analysis of several potentially co?administrated drugs with zonisamide. The stability of the analyte in blood specimen during sample handling was also verified by subjecting the samples to three freeze-thaw cycles, and up to 60 days maintenance at −40°. Stability of the solutions of zonisamide and the I.S. was studied over a period of 3 weeks by comparing the peak areas at different times.
The present analytical method was validated in a randomised crossover bioequivalence study. The study protocol was approved by Medical Ethics Committee of the university. Twenty four healthy volunteers aged 26.7±4.9 years and weighing 70.2±6.7 kg with normal biochemical parameters were divided into two groups. They received a single oral dose of 200 mg zonisamide from either Bakhtar Bioshimi (Kermanshah, Iran) or Eisai Ltd. (Zonergan; Spain) pharmaceutical companies after an overnight fasting. All the subjects were asked to refrain from food or water consumption for 3 h after drug administration. After 6 week wash?out period, the subjects were crossed?over. The blood samples were collected at suitable intervals up to 180 h after drug administration and stored frozen at 40° until analysis. Pharmacokinetic parameters including maximum concentration (Cmax), area under the concentration (AUC) time curve from zero to the time of last sampling (AUC 0?t) and AUC time curve from zero to infinity (AUC 0?∞) were calculated and compared using paired Student’s?test. Statistical significance was defined at the level of P<0.05.
Representative chromatograms of human blank serum spiked with the I.S. and zonisamide shown in figs. 1a and b, respectively (0.025 μg/ml). Under the chromatographic conditions described, no endogenous components from serum were found to interfere with the elution of zonisamide or the IS. Figs. 1c and d shows the chromatograms of serum samples obtained at 6 and 120 h after a single oral dose of 200 mg zonisamide from a healthy volunteer, respectively.

Figure 1: Typical chromatograms obtained from determination of zonisamide in human serum by the HPLC method.
(a) Human blank serum spiked with vanillin as the IS, (b) human blank serum spiked with 0.025 μg/ml zonisamide and vanillin, (c and d) serum samples obtained at 6 h and 120 h after a single oral dose of 200 mg zonisamide from a healthy volunteer containing 3.52 and 0.16 μg/ml of zonisamide, respectively. Peaks eluted at 2.2 and 2.9 min correspond to zonisamide and the IS, respectively
The standard calibration curves were linear over the concentration ranges of 0.015?6.4 μg/ml using line?fit plot in regression analysis with a coefficient of 0.9985 and regression equation of y=41.043x+2.948. Intra? and inter?day reproducibility for calibration curves were determined on the same day in replicate (n=6) and on different days (n=6), respectively, using same pooled serum sample. The intra?day average slope of the fitted straight lines was 41.043±2.452 μg/ ml (CV=5.9%) and the mean intercept of the calibration curves was 2.948±0.198 (CV=6.7%). The corresponding mean (±SD) coefficient of the linear regression analysis was 0.9985±0.0072 (CV=0.72%). For calibration curves prepared on different days, the mean±SD of results were as follows: Slope=40.092±3.152 μg/ml (CV=7.9%), coefficient of the linear regression analysis=0.9954±0.004 (CV=0.40%) and intercept=3.142±0.3466 (CV=11.0%).
The mean extraction efficacies of zonisamide and the IS from serum were found to be 101±1% and 87±5%, respectively. The inter and intra?day accuracy and precision values of the assay method have been presented in Table 1. The coefficient variation values of both intra? and interday analysis were less than 12.2% whereas the percentage error was found to be generally less than 4.4. The selectivity study was performed and using the same analytical conditions any peak was not detected from phenytoin, carbamazepine, topiramate, gabapentin, vigabatrine, oxcarbazepine, sodium valproate and diazepam up to 10 min. Ethosuxemide, lamotrigine, primidone, clonazepam and phenobarbital were eluted at 2.0, 3.6, 4.2, 4.4 and 8.4 min, respectively. Thus except for ethosuxemide there were no interfering peaks from other commonly co?administrated AEDs. Some other potentially co?administered drugs including: Diclofenac, theophylline, furosemide, acetaminophen, naproxen, indomethacin, celecoxib, propranolol, diltiazem, aspirin, salicylic acid, omeprazole, cimetidine, codeine and caffeine were checked for interference with zonisamide analysis and none of these drugs show interference with analysis of the drug. All the drugs were tested at concentrations range of 10?1000 ng/ml. The stock solutions of zonisamide and the IS were stable for at least 30 days when stored at 4°. The stability of the drug was found to be 100% from the initial value, after 60 days maintenance of the serum at −40° and following three thaw-freeze cycles.
| Known concentration (µg/ml) | Concentration found (mean±SD) | Coefficient of variation (%) | Accuracy (% mean deviation) |
|---|---|---|---|
| Inter?day | |||
| 0.015 | 0.143±0.0017 | 12.2 | −4.4 |
| 1.5 | 1.448±0.084 | 5.8 | −3.4 |
| 6.4 | 6.278±0.115 | 1.8 | −1.9 |
| Intra?day | |||
| 0.015 | 0.145±0.0012 | 8.4 | −3.3 |
| 1.5 | 1.460±0.099 | 6.8 | −2.6 |
| 6.4 | 6.291±0.137 | 2.2 | −1.7 |
Accuracy has been calculated as a mean deviation from the nominal values. HPLC=High performance liquid chromatography
Table 1: Inter?And Intra?Day Precision And Accuracy For Determination Of Zonisamide In Human Serum By The Hplc Method
The present method has successfully been used for the determination of the drug in a randomised crossover bioequivalence study following single oral administration of two different zonisamide preparations in 24 healthy volunteers. Typical serum concentration–time profile and the resulted pharmacokinetic parameters of the drug has been shown in fig. 2 and Table 2, respectively.
| Parameter/prep | Test | Reference | P value | |
|---|---|---|---|---|
| Tmax(h) | 3.65 | (1.82) | 3.92 (1.91) | NS |
| Cmax(µg/ml) | 3.75 | (1.41) | 3.55 (1.65) | NS |
| AUC 0?24 (µgh/ml) | 259.9 | (109.9) | 252.2 (121.2) | NS |
| AUC 0?∞ (µgh/ml) | 317.55 (146.4) | 304.9 (168.2) | NS | |
| T1/2(h) | 65.5 | (18.3) | 61.94 (19.7) | NS |
NS=No significant difference (P<0.05), Tmax=Time to maximum concentration, Cmax=Maximum concentration, AUC=Area under the concentration time curve, T1/2=Elimination half?life
Table 2: Pharmacokinetic Parameters Of Zonisamide In 24 Healthy Volunteers After Administration Of A Single 200 Mg Oral Dose Of The Drug (Mean±Sd)

Figure 2: Plasma profile of zonisamide after oral administration. Time profiles of zonisamide for two different preparations in 24 human volunteers after administration of a single 200 mg oral dose (mean serum concentrations) (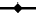 ) Test,
) Test, Reference
Reference
A few methods have been published for analysis of zonisamide in biological fluids. In the method described by Furuno et al. [21] detection limit of 0.1 μg/ml has been reported. This method is not sensitive enough to measure zonisamide concentrations up to three half?lives after a single dose administration and involves time consuming sample preparation procedure including back?wash extraction of the organic solvent. A sensitive method with LOQ of 0.05 μg/ml has been published for analysis of the drug in human breast milk and maternal plasma using solid?phase extraction [18]. In their method, however, long analytical run time (15 min) has been reported. A rapid method with analytical run time of about 5 min has been developed for analysis of zonisamide in human plasma using a nonporous silica column and solid phase extraction [22]. This method, however, is not enough sensitive (LOQ 1 μg/ml) to be used in human single?dose pharmacokinetic studies. Unlike time consuming and multi?steps extraction procedure which has been used in the previously published articles [15?18], a simple liquid-liquid extraction was developed in our study. The present article provides a new, fast and sensitive procedure for analysis of the drug in human serum. Our method which has been demonstrated to be suitable for use in human pharmacokinetic studies is fast with analytical run time of 3.5 min and sensitive (LOQ of 0.015 μg/ml) enough for measuring of the drug in single?dose bioequivalence studies of zonisamide, up to three half?lives post?dose.
Acknowledgements
This work was supported by Bakhtar Bioshimi Pharmaceutical Company and in part by Kermanshah University of Medical Sciences.
References
- Arzimanoglou A, Rahbani A. Zonisamide for the treatment of epilepsy. Expert Rev Neurother 2006;6:1283?92.
- Sackellares JC, Donofrio PD, Wagner JG, Abou?Khalil B, Berent S, Aasved?Hoyt K. Pilot study of zonisamide (1,2-benzisoxazole-3-methanesulfonamide) in patients with refractory partial seizures. Epilepsia 1985;26:206?11.
- Bermejo PE, Ruiz?Huete C, Anciones B. Zonisamide in managing impulse control disorders in Parkinson’s disease. J Neurol 2010;257:1682?5.
- Wilson MS, Findling RL. Zonisamide for bipolar depression. Expert OpinPharmacother 2007;8:111?3.
- Mohammadianinejad SE, Abbasi V, Sajedi SA, Majdinasab N, Abdollahi F, Hajmanouchehri R, et al. Zonisamide versus topiramate in migraine prophylaxis: A double?blind randomized clinical trial. Clin Neuropharmacol 2011;34:174?7.
- Biton V. Clinical pharmacology and mechanism of action of zonisamide. Clin Neuropharmacol 2007;30:230?40.
- Sills G, Brodie M. Pharmacokinetics and drug interactions with zonisamide. Epilepsia 2007;48:435?41.
- Wallace J, Shellenberger K, Groves L. Pharmacokinetics of zonisamide in young and elderly subjects. Epilepsia 1998;39:190?1.
- Baulac M. Introduction to zonisamide. Epilepsy Res 2006;68:S3?9.
- Subramanian M, Birnbaum AK, Remmel RP. High?speed simultaneous determination of nine antiepileptic drugs using liquid chromatography?mass spectrometry. Ther Drug Monit 2008;30:347?56.
- Gholivand MB, Khodadadian M, Majnooni MB, Bahrami GH. Electroreduction of zonisamide at hanging mercury drop electrode and its determination in pharmaceutical formulations and spiked human serum samples. Anal Lett 2010;43:269?79.
- Greiner?Sosanko E, Giannoutsos S, Lower DR, Virji MA, Krasowski MD. Drug monitoring: Simultaneous analysis of lamotrigine, oxcarbazepine, 10?hydroxycarbazepine, and zonisamide by HPLC?UV and a rapid GC method using a nitrogen?phosphorus detector for levetiracetam. J ChromatogrSci 2007;45:616?22.
- Kataoka Y, Makino K, Oishi R. Capillary electrophoresis for therapeutic drug monitoring of antiepileptics. Electrophoresis 1998;19:2856?60.
- Kalbe K, Nishimura S, Ishii H, Sunahara N, Naruto S, Kurooka S. Competitive binding enzyme immunoassay for zonisamide, a new antiepileptic drug, with selected paired?enzyme labeled antigen and antibody. Clin Chem 1990;36:24?7.
- Hosseini M, Alipour E, Farokhsir A. Determination and validation of zonisamide and its four related substances by HPLC and UV?spectrophotometry. Indian J Pharm Sci 2010;72:302?6.
- Rao DV, Chakravarthy IE, Kumar SR. Stability indication HPLC method for the determination of zonisamide as bulk drug and in pharmaceutical dosage form. Chromatographia 2006;64:261?6.
- Kim M, Tadashi N, Tsutomu SH, Kazuki T, Koichi Y, Tokenichi M. Evaluation of a highly sensitive measurement method of zonisamide by an HPLC system with column?switching. Jap J Pharm Health Care Sci 2003;29:178?83.
- Shimoyama R, Ohkubo T, Sugawara K. Monitoring of zonisamide in human breast milk and maternal plasma by solid?phase extraction HPLC method. Biomed Chromatogr 1999;13:370?2.
- Greiner?Sosanko E, Lower DR, Virji MA, Krasowski MD. Simultaneous determination of lamotrigine, zonisamide, and carbamazepine in human plasma by high?performance liquid chromatography. Biomed Chromatogr 2007;21:225?8.
- Yamashita S, Furuno K, Kawasaki H, Gomita Y, Yoshinaga H, Yamatogi Y, et al. Simple and rapid analysis of lamotrigine, a novel antiepileptic, in human serum by high?performance liquid chromatography using a solid?phase extraction technique. J Chromatogr B Biomed Appl 1995;670:354?7.
- Furuno K, Oishi R, Gomita Y, Eto K. Simple and sensitive assay of zonisamide in human serum by high?performance liquid chromatography using a solid?phase extraction technique. J Chromatogr B Biomed Appl 1994;656:456?9.
- Nakamura M, Hirade K, Sugiyama T, Katagiri Y. High?performance liquid chromatographic assay of zonisamide in human plasma using a non?porous silica column. J Chromatogr B Biomed SciAppl 2001;755:337?41.





