- *Corresponding Author:
- Joo Eun Kim
Department of Pharmaceutical Engineering, Catholic University of Daegu, Gyeongbuk 38430, Republic of Korea
E-mail: 77jooeun@naver.com
| Date of Received | 31 December 2019 |
| Date of Revision | 04 February 2021 |
| Date of Acceptance | 28 May 2021 |
| Indian J Pharm Sci 2021;83(3):451-464 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
Fixed dose complex dual-layer tablets of telmisartan and rosuvastatin calcium have been used for the treatment of hypertension and hyperlipidemia. However, the current United States Pharmacopoeia analysis method is limited by the need for twice the time and cost owing to analysis for each active pharmaceutical ingredient present separately. Due to the critical limitations in establishing simultaneous analysis methods for both active pharmaceutical ingredients, there is an urgent need for an analysis method that can be applied for immediate use by researchers. The objective of this study was to develop and validate a simultaneous quantitative assay using a rapid and selective high performance liquid chromatography method for the analysis of rosuvastatin calcium and telmisartan in fixed dose combination dual-layer tablet dosage forms. This method used Kinetex C18 (5 µm, 4.6×150 mm) columns at a flow rate of 1.0 ml/ min and Ultraviolet-visible detector set at a wavelength of 242 nm. The separation was carried out using an ammonium phosphate monobasic buffer (pH 3.0) and methanol at a ratio of 300:700 as the mobile phase. The retention times of rosuvastatin calcium and telmisartan were 2.2 and 4.3 min respectively. The system suitability was 0.41 % for rosuvastatin calcium and 0.63 % for telmisartan, and the peak of each component showed complete separation from disturbance peaks. The correlation coefficient of the active pharmaceutical ingredients was approximately 0.999 when the concentration of rosuvastatin calcium was in the range of 4.44-26.6 µg/ml and the concentration of telmisartan was 0.176-106.6 µg/ml, this indicates good linearity. The recovery rate relative to the amount added was 97.0–99.6 % for rosuvastatin calcium and 95.5-102.2 % for telmisartan. Precision and solution stability were within the permissible range. Validation acceptance criteria were met in all cases. This simultaneous quantitative analysis method was successfully applied to the quality assessment of six fixed dose combination dual-layer tablets containing various amounts of rosuvastatin calcium and telmisartan.
Keywords
Telmisartan, rosuvastatin calcium, simultaneous estimation, method validation, dissolution, high performance liquid chromatography
Rosuvastatin calcium (bis[(E)-7-[4-(4-fluorophenyl)- 6-isopropyl]-2-[methyl](methylsulfonyl)amino] pyrimidin-5-dihydroxyhept-6-enoic acid] calcium salt) is used to reduce cholesterol in hyperlipidemic patients by inhibiting the synthesis of cholesterol[1-3]. Telmisartan (2-(4-{[4-methyl-2-propyl-1H-1,3- benzodiazol-1-yl]methyl}phenyl)benzoic acid) blocks the action of angiotensin II (a substance that increases the body’s water content) to expand blood vessels and reduce the body’s water content, which can lower the risk of cardiovascular disease[4-6]. Recently, these two drugs have been developed into a fixed-dose combination (FDC) drug. A complex bilayer tablet of telmisartan and rosuvastatin calcium was developed to increase the convenience of treating patients for hypertension/hyperlipidemia and to obtain a synergistic effect in terms of treatment by drug interaction. The commercially available telmisartan and rosuvastatin calcium complex bilayer tablets are called “Duowell” and are available in six different dosages (80/20, 80/10, 80/5, 40/20, 40/10 and 40/5 mg). In the development of pharmaceuticals, analysis of pharmaceutical is a criterion for evaluating manufactured pharmaceuticals and an optimized analysis method is required for accurate evaluation. Multiple High Performance Liquid Chromatography (HPLC)-based methods are available for both individual compounds and combinations. However, individual analysis methods of the United States Pharmacopoeia for measuring telmisartan and rosuvastatin are time-consuming and costly in the pharmaceutical industry field. Further, if both drugs are analyzed simultaneously, the interference of various excipients affects these analyses. In addition, analyzing each active pharmaceutical ingredient individually in the analysis of complex dual-layer tablets takes a long time for quality control (QC), and it is expensive to evaluate all six doses[7-9].
Therefore, our research team applied several simultaneous analysis methods currently developed to analyze the telmisartan/rosuvastatin calcium complex dual-layer tablets. However, due to the physicochemical properties of the active pharmaceutical ingredients and excipients it was difficult to establish specificity in the analysis method for the complex FDC formulation. Moreover, it is not possible to secure linearity to be compatible with the range of all six dosage forms. Therefore, for the accurate and precise quality evaluation of the developed composite formulation, the development of a new analysis method is required. The aims of this study were to develop a method capable of simultaneous analysis of telmisartan and rosuvastatin calcium and to verify the validity of the developed analysis method. The analysis method applicable from low to high dose in the range of six dosage formulations allows the simultaneous analysis of all six doses in one analysis process. In this way, the analysis time and cost can be reduced by over half.
Currently, there is no Reversed-phase High Performance Liquid Chromatography (RP-HPLC) method for the simultaneous quantitative analysis of rosuvastatin calcium and telmisartan. Furthermore, there is no RP-HPLC method for simultaneously estimating the dissolution of telmisartan and rosuvastatin calcium in dual-layer tablet dosage forms. Therefore, we developed a new quantitative analysis method for rosuvastatin calcium and telmisartan in a dissolution test solution. We validated the method by testing its system suitability, specificity, linearity, accuracy and precision, limit of quantitation and detection and solution stability.
Materials and Methods
Materials:
Certified reference standards of rosuvastatin calcium and telmisartan were provided by the USP (Rockville, MD, USA). Rosuvastatin calcium was provided by Melody Healthcare, Ltd. (Mumbai, India). Telmisartan was provided by Dongbang FTL Ltd. (Seoul, Korea). d-Mannitol 200 SD and d-mannitol 100 SD were provided by Roquette PTE Ltd. (Singapore, Singapore). MicroceLac 100 was provided by Meggle Pharma (Wasserburg, Germany). Aerosil was provided by Evonik Industries AG (Essen, Germany). Red iron oxide was provided by Univar Colour (Billericay, UK). Crospovidone and povidone were provided by BASF (Ludwigshafen, Germany).Meglumine was provided by Suzhou Tianma Specialty Chemicals Co., Ltd. (Jiangsu, China). Microcrystalline cellulose and sodium stearyl fumarate were provided by JRS Pharma (Patterson, NY, USA). Magnesium stearate was provided by Nitika Pharmaceutical Specialties Pvt. Ltd. (Maharashtra, India). Liquid chromatography-grade ethanol, EP-grade phosphate, ammonium phosphate monobasic and ultra-pure water were used to make the mobile phase.
Preparation of the pharmaceutical formulations:
The telmisartan layer consisted of 40-80 mg of telmisartan, with 287.28 mg of Pearlitol SD 100 (Roquette Ltd., Lestrem, France), 45.2 mg of Pearlitol SD 200 (Roquette Ltd.), 6.72 mg of sodium hydroxide (Sigma-Aldrich, St. Louis, MO, USA), 24 mg of meglumine (Suzhou Tianma Specialty Chemicals Co., Ltd.), 24 mg of povidone (JRS Pharma Co., Ltd.), 7.2 mg of sodium stearyl fumarate (JRS Pharma Co., Ltd.) and 5.6 mg of magnesium stearate (Nitika Pharmaceutical Specialities Pvt., Ltd.) added as excipients. The rosuvastatin calcium layer was composed of 5-20 mg rosuvastatin calcium and 39 mg of crospovidone (JRS Pharma Co., Ltd.), 20 mg of calcium glycerophosphate (Isaltis SAS, Lyon, France), 213.64 mg of Microcelac 100 (Meggle Pharma), 0.06 mg of iron oxide red (Colorcon Inc., Harleysville, PA, USA), 2.5 mg of magnesium stearate and 4 mg of Aerosil 200 (Evonik Industries AG) were added as excipients. The telmisartan layer consisted of wet granules using fluid bed granulation (GPCG 1; Glatt, Binzen, Germany). The rosuvastatin calcium layer resulted from direct blending only, and it was finally blended with magnesium stearate. Dual-layered telmisartan-rosuvastatin tablets were prepared by compression using a rotary tablet press machine for dual-layer and coating with a film coating machine to a targeted hardness and mass weight of 8-11 kPa and 440-780 mg respectively.
Method development:
Information on the physicochemical properties of the raw materials of rosuvastatin calcium and telmisartan free base was collected and a detection mode suitable for analysis was determined. When testing the dual-layer tablets of rosuvastatin and telmisartan, the peaks derived from each analysis method were confirmed after applying the conventional separate analysis method of US Pharmacopoeia (USP) 42-NF37 among HPLC conditions. It was confirmed that the placebo peak overlapped the active pharmaceutical ingredient (API) peak with the main component peak and a forced decomposition test was also conducted. Three sample solutions suggested in USP42-NF37 were prepared and a comparative test was conducted. For verification, each recovery rate relative standard deviation (RSD) %, T-value, F-value and specificity between USP42-NF37 and the developed method confirmed the possibility of simultaneous analysis.
Dissolution:
To prepare a pH 1.2 dissolution test solution, 0.7 ml of 35 % HCl and 200 mg of NaCl were accurately measured, taken in a beaker and dissolved in 1000 ml of deionized water. To prepare a pH 4.0 dissolution test solution, 0.246 ml of glacial acetic acid and 73.8 mg of sodium acetate anhydrous were accurately measured, taken in a beaker and dissolved in 1000 ml of deionized water. To prepare a pH 6.8 dissolution test solution, 6.805 g of potassium dihydrogen phosphate and 0.8 g of NaOH were accurately measured taken in a beaker and dissolved in 1000 ml of deionized water. Using a dissolution testing machine (Varian VK7020, Agilent Instruments, Santa Clara, CA, USA), all experiments were conducted under the following USP <711> dissolution conditions. The in vitro dissolution study was conducted in 900 ml of USP dissolution solutions (pH 1.2, 4.0 and 6.8 and water) in a 1000 ml vessel maintained at 37±0.5°. The telmisartan and rosuvastatin calcium dual-layer tablets were added to the containers of a type II (paddle method, USP23) dissolution apparatus and operated at 50±0.02 rpm. Dissolution sampling was done at 5, 10, 15, 30, 45, 60, 90 and 120 min. At each time point, a 5 ml sample was collected using an autosampler and filtered through an RC filter (0.45 µm, 25 mm) to create a dissolution sample, which was analyzed by HPLC. The percentage of rosuvastatin calcium and telmisartan dissolved in the samples was calculated by comparing the measured peak areas of the dissolution test samples and the USP standard.
The following equation was used:
Dissolved (%) = [(900 ml/drug loading rate)×(peak area (sample)]/[peak area (USP standard)×concentration of the USP standard]×100.
In addition, we compared the dissolution profile of our developed tablets and commercial products. In the APIs of telmisartan and rosuvastatin each, the similarity factors (F2) were evaluated at pH 1.2, pH 4.0 and pH 6.8, and in water as the dissolution test solution.
The following equation was used:
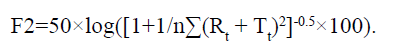
Chromatographic conditions (RP-HPLC):
The RP-HPLC system comprised a Waters HPLC system (Waters 1529, UV/Vis 2707; Waters, Milford, MA, USA) equipped with a binary pump, column heater, auto sampler and Ultraviolet-Visible (UV/Vis) detector. The data collection and analysis for the simultaneous measurement of rosuvastatin calcium and telmisartan were performed using Empower 3 software (Waters). A Kinetex C18 (5 μm, 4.6×150 mm) column was used and the liquid chromatography (LC) detector UV/Vis detector was set at a wavelength of 242 nm to analyze rosuvastatin calcium and telmisartan[10-17]. The separation was performed using a mobile phase comprising ammonium phosphate monobasic buffer (pH 3.0) and methanol at a ratio of 300:700. The flow rate was 1.0 ml/min. The injection volume was set to 10 µl, the run time was set to 11 min and the column temperature was set to 40°.
Preparation of the standard solutions and QC samples:
Preparation of the standard stock solutions: To generate the rosuvastatin calcium USP reference standard, exactly 11.6 mg of rosuvastatin calcium and 25 ml of water were combined in a 50 ml volumetric flask and sonicated for 10 min. Next, 12.5 ml of acetonitrile was added and the total volume was brought up to 50 ml with water. The final concentration of rosuvastatin calcium was 222 µg/ml. To generate the telmisartan USP reference standard, exactly 44.4 mg of telmisartan and 50 mL of methanol were combined in a 50 ml volumetric flask. The final concentration of telmisartan was 888 µg/ml.
Preparation of the standard solutions: Standard solutions of rosuvastatin calcium with pH 1.2 and 4.0 were prepared by diluting 5 ml of the rosuvastatin stock solution with the pH 1.2 and 4.0 dissolution media, respectively, in a 50 ml volumetric flask to reach a final concentration of 22.2 µg/ml. To prepare the rosuvastatin calcium standard solution with pH 6.8 or water, 5 ml of the rosuvastatin stock solution was diluted with the pH 6.8 dissolution media or water in a 50 ml volumetric flask. The resulting solution was then diluted 1:1 with methanol to achieve a final concentration of 11.1 µg/ml rosuvastatin calcium.
The standard solution of telmisartan with a pH of 1.2 was prepared by diluting 5 ml of the telmisartan stock solution with the pH 1.2 dissolution media in a 50 ml volumetric flask to reach a final concentration of 88.8 µg/ml. Since telmisartan displayed low solubility in the pH 4.0 dissolution media, 2 ml of the telmisartan stock solution was diluted with the pH 4.0 dissolution media in a 100 ml volumetric flask to reach a final concentration of 17.8 µg/ml. Since telmisartan is physicochemically unstable at pH 6.8 and in water, it was diluted with the dissolution solutions after pretreatment with methanol. To prepare the telmisartan standard solution with pH 6.8 or water, 5 ml of the telmisartan stock solution was diluted in a 50 ml volumetric flask with methanol. The resulting solution was then diluted 1:1 with the pH 6.8 dissolution media or water to achieve a final concentration of 44.4 µg/ml telmisartan.
Preparation of the QC samples:
Following the paddle method of testing dissolution described in the US Pharmacopoeia <711>, six tablets of telmisartan-rosuvastatin calcium were tested in six individual vessels (1 tablet per vessel) with 900 ml of each dissolution test solution at 37±0.5° and 50 rpm. The dissolution solution in which the tablets were dissolved was filtered until it reached maximum concentration. After the dissolution test, 10 ml was taken from the vessel and filtered. The filtered solution was used as the QC test solution. QC samples containing pH 1.2 and 4.0 media were not diluted, however QC samples containing pH 6.8 media or water were diluted with methanol at a ratio of 1:1.
Method validation:
The method was validated according to USP category I requirements. The following validation characteristics were addressed: linearity, range, accuracy, precision, limit of quantitation, specificity, and solution stability[18-25] .
System suitability:
The system suitability of rosuvastatin calcium and telmisartan each was confirmed by measuring the peak area produced by the repeated injection of the standard solution in each dissolution test solution at pH 1.2, 4.0 or 6.8 and water[26-28]. The system suitability standard solution which contained 22.2 µg/ml rosuvastatin calcium and 88.8 µg/ml telmisartan was prepared by diluting and mixing rosuvastatin calcium and telmisartan stock solutions with four dissolution test solutions. Because of the low solubility of telmisartan at pH 4.0, the system suitability standard solution containing 17.8 µg/ml of telmisartan was prepared by diluting and mixing stock solutions with the dissolution medium. System suitability was determined from six repeated injections of the system suitability standard before sample analysis. For each of the four tests, the acceptance criteria were a relative standard deviation of ≤1.0 % for peak area, a USP tailing factor of ≤ 2.0 (tailing factor ≤ 1.5 for rosuvastatin calcium) and theoretical plates (N) ≥2000.
Specificity:
Specificity was determined by analyzing samples containing a mixture of the drug product excipients as well as samples containing the main degradation products of rosuvastatin calcium and telmisartan. All chromatograms were examined to determine if rosuvastatin calcium, telmisartan, and their degradation products eluted with each other or with any excipient peaks. Excipients included sodium hydroxide, povidone, meglumine, d-mannitol, stearyl sodium fumarate, magnesium stearate, microcellac 100, calcium glycerophosphate, crospovidone, red iron oxide, colloidal silicon dioxide, hypromellose, titanium dioxide, polyethylene glycol 400, talc, and polyethylene glycol 6000. Blank, placebo, standard, and sample solutions were prepared and analyzed by RP-HPLC according to the dissolution test solution used (i.e., pH 1.2, 4.0, or 6.8, or water). The two APIs should be free from interference and should be separate from interference peaks.
Linearity and range:
To confirm linearity in the four dissolution test solutions, a standard calibration curve for rosuvastatin calcium was prepared with five calibrators over a concentration range of 4.44-26.6 µg/ml (prepared with 20 %, 40 %, 80 %, 100 % and 120 % of the maximum dissolution rate). For telmisartan, seven calibrators were prepared over a concentration range of 0.176-44.4 µg/ml (prepared with 0.2 %, 1 %, 10 %, 20 %, 50 %, 100 % and 120 % of the maximum dissolution rate; 0.2-50 % with the pH 4.0 medium). The peak area versus drug concentration data was analyzed by linear least-squares regression and the standard curves were evaluated for linearity. The range was the interval between the highest and lowest concentration of the analyte where acceptable linearity, accuracy and precision were obtained. Because rosuvastatin calcium and telmisartan tablets consist of six dose forms (80/20, 80/5, 80/10, 40/20, 40/5 and 40/10 mg), the concentration range was set to enable simultaneous quantitative analysis from a high dose to a low dose. Minitab® software (version 18; Minitab Inc., State College, PA, USA) was used for statistical evaluation of linearity. To identify the residual plot of linearity, the error range for each pH was evaluated by setting the X value of linearity as the input value. Through this, the range of the residual was identified. Regression equations and variance analysis were used to determine the suitability of linearity.
Accuracy and precision:
Accuracy and precision were determined by analyzing QC standard samples at three concentrations of rosuvastatin calcium and telmisartan (40 %, 100 % and 120 % of telmisartan [88.8 µg/ml] and rosuvastatin [23.1 µg/ml]). For the pH 4.0 dissolution test solution, the concentration was prepared considering the solubility of telmisartan (10 %, 20 % and 50 % of telmisartan [88.8 µg/l). The accuracy was evaluated by using three concentration levels within the calibration range and measuring the percent recovery rate compared to the amount added. Accuracy was established by evaluating the amount determined from the QC standards and comparing it to the respective nominal value expressed as percent recovery. Precision was expressed by the percentage R.S.D. of the analyte peaks. The precision was measured by repeating the preparation of the QC samples at 40 %, 100 %, 120 % concentration levels for 2 non-consecutive days, using another set of test equipment and different testers. The approval criteria for the analytical methods established in accordance with the International Council for Harmonisation (ICH) Guidelines and Medicinal Products Test Methods Validation Guidelines (revised version) are 100 % ± 5 % accuracy at recovery and 5 % or less relative standard deviation for recovery.
For a found concentration, accuracy and precision can be determined using the following equations: Found concentration (µg/ml=(QC sample peak area-intercept)/slope, Accuracy=(found concentration/spiked concentration)×100, Precision (RSD)=(SD of found concentrations/average of found concentrations)×100.
Limit of quantitation and detection:
The limits of quantitation for rosuvastatin calcium and telmisartan were defined as the lowest concentration where acceptable accuracy and precision were obtained. The limit of detection is defined as the concentration at which the main ingredient sample is detectable. A simultaneous quantitative analysis method was used to determine the minimum concentrations of rosuvastatin calcium and telmisartan available for analysis. In addition, an estimation of the limit of quantitation for rosuvastatin calcium and telmisartan was calculated from 10 times the signal to noise value and the limit of detection was calculated from 3.3 times the signal to noise value. The limit of quantitation (S/N=10) and limit of detection (S/N=3.3) were acquired based on the SD of the y-intercept and the slope using the results of the linearity test repeated three times.
Robustness:
The robustness of the method was evaluated by analyzing the system suitability standard and evaluating system suitability parameter data after varying the HPLC pump flow rate (±10 %), the auto-sampler injector volume (±50 %), and the column compartment temperature (±4°), individually[29, 30].
Solution stability:
To confirm the solution stability of the two APIs in the four dissolution media, the stability between the standard samples containing the USP rosuvastatin calcium reference standard and the USP telmisartan reference standard were compared with the reference samples produced with the rosuvastatin/telmisartan FDC tablets in the four dissolution test solutions. The period of stability was set at 24 h and 30 h for the two APIs. An acceptable standard criterion was set to <5 % for the solution stability over time.
Results and Discussion
The chemical structures of rosuvastatin calcium and telmisartan are shown in fig. 1. Through preliminary study, it was confirmed that both APIs are non-polar drugs and have a common peak measurement range of 235-245 nm due to structural characteristics. The amount of rosuvastatin calcium and telmisartan in the dissolution sample was analyzed and validated using simultaneous quantitative analysis with RP-HPLC. Kinetex C18 (5 µm, 4.6×150 mm) columns were used at a flow rate of 1.0 ml/min, and the UV absorption was measured at a wavelength of 242 nm. The separation was performed using a mobile phase comprising ammonium phosphate monobasic buffer (pH 3.0) and methanol. It was prepared by dissolving 2.0 g of ammonium dihydrate phosphate in distilled water to make the volume 1 l and then 300 ml of ammonium buffer was added to adjust the pH to 3.0. The ammonium phosphate monobasic buffer was then mixed with methanol at a ratio of 300:700. The retention times of rosuvastatin calcium and telmisartan were 2.2 and 4.3 min, respectively.

Figure 1: The chemical structure of telmisartan(A), and rosuvastatin calcium(B)
The standard deviation of the system suitability test results for four dissolution test solutions was 0.63 % or less for rosuvastatin calcium and telmisartan. The rosuvastatin calcium dissolution condition with pH 1.2 satisfied the suitable standard. The relative standard deviation of the summation of peak areas of rosuvastatin calcium and its degradation products was 0.30 % (Table1).
| Test number | Rosuvastatin calcium | Telmisartan | ||||||
|---|---|---|---|---|---|---|---|---|
| pH1.2a | pH4.0 | pH6.8 | Water | pH1.2 | pH4.0 | pH6.8 | Water | |
| 1 | 590114 | 555607 | 285386 | 290137 | 3298151 | 633529 | 1748062 | 1725421 |
| 2 | 589667 | 553623 | 287341 | 288886 | 3287942 | 633716 | 1762175 | 1731177 |
| 3 | 589806 | 553355 | 288490 | 287724 | 3305703 | 633589 | 1759858 | 1728844 |
| 4 | 587287 | 554956 | 287137 | 290567 | 3305711 | 635015 | 1738528 | 1723939 |
| 5 | 586299 | 554001 | 287601 | 290607 | 3315304 | 634511 | 1744114 | 1727134 |
| 6 | 586555 | 554004 | 288285 | 290571 | 3315472 | 633243 | 1735051 | 1714723 |
| Mean | 588288 | 554257.7 | 287373.3 | 289748.7 | 3304714 | 633933.8 | 1747965 | 1725206 |
| RSD(%) | 0.3 | 0.1 | 0.3 | 0.4 | 0.3 | 0.1 | 0.6 | 0.3 |
aThe summation of rosuvastatin and its degradant peaks
Table 1: Results of Injection Repeatability for Rosuvastatin Calcium and Telmisartan in Four Dissolution Medium (Medium : Ph1.2, Ph4.0, Ph6.8, Water)
As shown in fig. 2, blank, placebo, standard and sample solutions were prepared for each dissolution test solution to confirm specificity. Interference peaks were not observed for rosuvastatin calcium and telmisartan (fig. 2B, 2C and 2D). Rosuvastatin calcium was hydrolyzed at pH 1.2 to form a decomposition product. However, the purpose of this study is to confirm the presence or absence of peak interference between the APIs and the excipients. Therefore, the peak generated by the decomposition of rosuvastatin calcium in the dissolution solution of pH 1.2 did not affect the excipient peak, and as a result, the specificity of the developed method was secured (fig. 2A).

Figure 2: HPLC chromatograms demonstrating the selectivity of the analytical method for the determination of rosuvastatin calcium and telmisartan (Medium: pH 1.2(A), pH 4.0(B), pH 6.8(C), and water(D))
For the four dissolution test solutions, linearity was investigated by preparing a rosuvastatin calcium standard curve in the analytical range of 20-120 % (4.44-26.6 µg/ml) (Table 2) and a telmisartan standard curve in the analytical range of 0.2-120 % (0.176-106.6 µg/ml). For the pH 4.0 dissolution medium, the range was 0.2-50 % (0.176-44.4 µg/ml) (Table 3). As shown in fig. 3 and 4, excellent correlation with the calibration curve was observed with a correlation coefficient (R2) of 0.999 for all standard curves. The validation concentrations at 80-120 % were not tested because the concentration is considered for the range of all formulations from high to low concentrations. As shown in Table 2 and 3, method validation was performed on all ranges of 80/20, 80/10, 80/5, 40/20, 40/10 and 40/5 mg in the rosuvastatin and telmisartan FDC tablets. Thus, all were validated to enable quantitative analysis and simultaneous estimation (fig. 2, 3 and 4). Linear regression analysis was performed for each dissolution test solution and the following linear regression equation was obtained. For telmisartan, we obtained linear regression equations of y=32955x+1348.7 at pH 1.2, y=31566x+17.403 at pH 4.0, y=17864x−537.57 at pH 6.8, and y=17272x+696.69 in water. Further, for rosuvastatin calcium, we obtained linear regression equations of y=5838.1x+2703.9 at pH 1.2, y=5558.6x–1859.9 at pH 4.0, y=2844.9x+2090.2 at pH 6.8, and y=2849x+1079.3 in water.

Figure 3: The linearity of the calibration curve for telmisartan (Medium: pH1.2, pH4.0, pH6.8, water) LC conditions: C18 (5 µm, 4.6 x 150 mm) column, detector 242 nm, run time 11minute, column temperature 40°C

Figure 4: The linearity of the calibration curve for rosuvastatin calcium (Medium: pH1.2, pH4.0, pH6.8, water) LC conditions: C18 (5 µm, 4.6 x 150 mm) column, detector 242 nm, run time 11minute, column temperature 40°C
| pH1.2 | |||
| Concentration level (%) | peak areaa | ||
| test1 | test2 | test3 | |
| 20 | 119554 | 118523 | 118834 |
| 40 | 233433 | 233649 | 233233 |
| 80 | 474732 | 467555 | 466942 |
| 100 | 587339 | 581159 | 580385 |
| 120 | 700193 | 699323 | 698985 |
| slope | 27206.65 | 27049.23 | 26995.05 |
| Y-intercept | 2708.88 | 2060.53 | 2172.37 |
| r2 | 0.999 | 0.999 | 0.999 |
| pH4.0 | |||
| Concentration level (%) | peak area | ||
| test1 | test2 | test3 | |
| 20 | 112816 | 113157 | 114381 |
| 40 | 217561 | 219409 | 220707 |
| 80 | 441434 | 442883 | 442795 |
| 100 | 550946 | 552735 | 552732 |
| 120 | 669029 | 667294 | 669479 |
| slope | 25884.03 | 25826.28 | 25818.87 |
| Y-intercept | -1859.91 | -91.07 | 843.56 |
| r2 | 0.999 | 0.999 | 0.999 |
| pH6.8 | |||
| Concentration level (%) | peak area | ||
| test1 | test2 | test3 | |
| 20 | 59083 | 57952 | 58488 |
| 40 | 115370 | 114934 | 115410 |
| 80 | 230174 | 229786 | 228105 |
| 100 | 287199 | 288602 | 287317 |
| 120 | 342797 | 343659 | 342212 |
| slope | 13206.73 | 13310.44 | 13196.59 |
| Y-intercept | 2090.21 | 579.09 | 1594.12 |
| r2 | 0.999 | 0.999 | 0.999 |
| Water | |||
| Concentration level (%) | peak area | ||
| test1 | test2 | test3 | |
| 20 | 56981 | 57649 | 57301 |
| 40 | 115556 | 114199 | 114292 |
| 80 | 228937 | 225696 | 225971 |
| 100 | 289405 | 283850 | 285838 |
| 120 | 340141 | 343967 | 340837 |
| slope | 13225.44 | 13235.05 | 13178.38 |
| Y-intercept | 1079.35 | -166.19 | 417.91 |
| r2 | 0.999 | 0.999 | 0.999 |
aThe summation of rosuvastatin and its degradant peaks
Table 2: The Results of Linearity Test for Rosuvastatin Calcium (Medium : Ph1.2, Ph4.0, Ph6.8, Water)
| pH1.2 | |||
| Concentration level (%) | peak area | ||
| test1 | test2 | test3 | |
| 0.2 | 4923 | 5517 | 5083 |
| 1 | 32732 | 32338 | 33473 |
| 10 | 332556 | 333604 | 334519 |
| 20 | 661882 | 665494 | 659634 |
| 50 | 1652710 | 1660862 | 1664432 |
| 100 | 3295384 | 3317039 | 3330201 |
| 120 | 3955324 | 3972595 | 3981271 |
| slope | 37278.76 | 37475.75 | 37598.06 |
| Y-intercept | 1348.69 | 1339.41 | -160.74 |
| r2 | 0.999 | 0.999 | 0.999 |
| pH4.0 | |||
| Concentration level (%) | peak area | ||
| test1 | test2 | test3 | |
| 0.2 | 5408 | 5690 | 5231 |
| 1 | 31126 | 30326 | 29155 |
| 5 | 149475 | 152231 | 152069 |
| 10 | 323286 | 326647 | 322874 |
| 20 | 636032 | 638867 | 631479 |
| 50 | 1575771 | 1659018 | 1589068 |
| slope | 35683.39 | 37552.17 | 35971.62 |
| Y-intercept | 17.4 | -8399 | -2172.46 |
| r2 | 0.999 | 0.999 | 0.999 |
| pH6.8 (Dilute with methanol) | |||
| Concentration level (%) | peak area | ||
| test1 | test2 | test3 | |
| 0.2 | 4320 | 2762 | 4422 |
| 1 | 18989 | 18405 | 18836 |
| 10 | 178757 | 179331 | 180445 |
| 20 | 355596 | 360103 | 358611 |
| 50 | 888761 | 888551 | 890857 |
| 100 | 1784275 | 1783553 | 1783816 |
| 120 | 2146323 | 2128452 | 2131496 |
| slope | 20217.4 | 20100.62 | 20113.33 |
| Y-intercept | -537.57 | 1515.53 | 2044.49 |
| r2 | 0.999 | 0.999 | 0.999 |
| Water (Dilute with methanol) | |||
| Concentration level (%) | peak area | ||
| test1 | test2 | test3 | |
| 0.2 | 3565 | 4268 | 2857 |
| 1 | 17926 | 17731 | 16732 |
| 10 | 176051 | 173216 | 173370 |
| 20 | 346382 | 346413 | 347487 |
| 50 | 859818 | 863015 | 858845 |
| 100 | 1731672 | 1737469 | 1729345 |
| 120 | 2071824 | 2088505 | 2071558 |
| slope | 19547 | 19677.72 | 19541.77 |
| Y-intercept | 696.69 | -1051.26 | -261.26 |
| r2 | 0.999 | 0.999 | 0.999 |
Table 3: Results of Linearity Test for Telmisartan (Medium : Ph1.2, Ph4.0, Ph6.8, Water)
The peak area for each pH in the concentration ranges was used to obtain standardized residuals through regression equations and variance analysis by using Minitab. In the case of rosuvastatin calcium, the R2 value was estimated to be 99.99% at pH 1.2, 99.98% at pH 4.0, 99.99% at pH 6.8, and 99.97 % in water. In the case of telmisartan, the R2 value was estimated to be 100 % at pH 1.2, 99.92 % at pH 4.0, 100 % at pH 6.8 and 100 % in water. The p value was also higher than 0.05, thus the statistical evaluation was deemed appropriate (fig. 5).

Figure 5: Plots of standardized residuals of rosuvastatin calcium(A), Plots of standardized residuals of telmisartan (B)
Accuracy and precision were established across the analytical range for rosuvastatin calcium and telmisartan and calculated with the QC samples. The accuracy and precision results of rosuvastatin calcium and telmisartan are summarized in Tables 4 and 5 respectively. The accuracy results for rosuvastatin calcium and telmisartan in all dissolution media showed good recovery. The accuracy of rosuvastatin calcium and telmisartan tested in drug products at three concentration levels obtained by the addition of a USP reference standard ranged from 97.0 % to 99.6 % and 95.5 % to 102.2 %, respectively for all dissolution test solutions. Results for the precision of rosuvastatin calcium and telmisartan ranged from 0.81 % to 1.36 % and 0.45 % to 1.98 % respectively for all dissolution test solutions.
| Label(Level) | Spiked conc.(ug/mL) | Found conc.(ug/mL)a | Accuracy*(%)b | Average(%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pH 1.2 | QC-1(40%) | 8.583 | 8.247 8.281 8.292 |
96.1 96.5 96.6 |
96.4 | |||||
| QC-2(100%) | 21.46 | 21.31 21.28 21.23 |
99.3 99.2 99.0 |
99.1 | ||||||
| QC-3(120%) | 25.76 | 25.39 25.44 25.55 |
98.6 98.8 99.2 |
98.9 | ||||||
| Total average(%) | 98.1 | |||||||||
| Total RSD(%) | 1.3 | |||||||||
| pH 4.0 | QC-1(40%) | 8.590 | 8.275 8.313 8.302 |
96.3 96.8 96.7 |
96.6 | |||||
| QC-2(100%) | 21.48 | 21.14 20.68 21.16 |
98.4 96.3 98.5 |
97.8 | ||||||
| QC-3(120%) | 25.77 | 24.91 24.95 24.86 |
96.7 96.8 96.5 |
96.7 | ||||||
| Total average(%) | 97.0 | |||||||||
| Total RSD(%) | 0.8 | |||||||||
| pH 6.8 | QC-1(40%) | 8.617 | 8.470 8.515 8.498 |
98.3 98.8 98.6 |
98.6 | |||||
| QC-2(100%) | 21.54 | 21.67 21.76 21.69 |
100.6 101.0 100.7 |
100.8 | ||||||
| QC-3(120%) | 25.85 | 25.89 25.75 25.56 |
100.2 99.6 98.9 |
99.6 | ||||||
| Total average(%) | 99.6 | |||||||||
| Total RSD(%) | 1.0 | |||||||||
| Water | QC-1(40%) | 8.617 | 8.578 8.628 8.452 |
99.6 100.1 98.1 |
99.3 | |||||
| QC-2(100%) | 21.54 | 21.47 21.61 21.62 |
99.6 100.3 100.4 |
100.1 | ||||||
| QC-3(120%) | 25.85 | 25.54 25.48 25.69 |
98.8 98.6 99.4 |
98.9 | ||||||
| Total average(%)` | 99.4 | |||||||||
| Total RSD(%) | 0.8 | |||||||||
Abbreviation : QC mean Quality Control Sample, RSD mean Relative Standard Deviation, aFound conc.(ug/mL) : (QC sample peak area ? intercept) / slope, bAccuracy : Found conc. / Spiked conc. x 100, cPrecision(RSD) : SD(Found concs,) / Average(Found concs.) x 100, *Calculated from the summation of rosuvastatin and its degradant peaks
Table 4: Results of Accuracy and Precision Test for Rosuvastatin Calcium (Medium : Ph1.2, Ph4.0, Ph6.8, Water)
| Label(Level) | Spiked conc.(ug/mL) | Found conc.(ug/mL)a | Accuracy*(%)b | Average(%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pH 1.2 | QC-1(40%) | 17.68 | 17.90 17.83 17.63 |
101.2 100.8 99.7 |
100.6 | |||||
| QC-2(100%) | 88.40 | 89.48 89.13 89.36 |
101.2 100.8 101.1 |
101.0 | ||||||
| QC-3(120%) | 106.1 | 107.0 107.2 107.0 |
100.9 101.0 100.9 |
100.9 | ||||||
| Total average(%) | 100.9 | |||||||||
| Total RSD(%) | 0.4 | |||||||||
| pH4.0 | QC-1(10%) | 8.846 | 9.226 9.202 9.271 |
104.3 104.0 104.8 |
104.4 | |||||
| QC-2(20%) | 17.69 | 18.21 17.84 18.09 |
102.9 100.8 102.2 |
102.0 | ||||||
| QC-3(50%) | 44.23 | 44.64 44.29 44.20 |
100.9 100.1 99.9 |
100.3 | ||||||
| Total average(%) | 102.2 | |||||||||
| Total RSD(%) | 1.8 | |||||||||
| pH6.8 | QC-1(40%) | 17.67 | 16.73 16.73 16.19 |
94.7 94.7 91.6 |
93.7 | |||||
| QC-2(100%) | 88.36 | 86.25 84.88 84.08 |
97.6 96.1 95.1 |
96.3 | ||||||
| QC-3(120%) | 106.0 | 104.0 102.0 100.9 |
98.1 96.2 95.1 |
96.5 | ||||||
| Total average(%) | 95.5 | |||||||||
| Total RSD(%) | 1.9 | |||||||||
| Water | QC-1(40%) | 17.67 | 17.45 17.48 17.51 |
98.8 98.9 99.1 |
98.9 | |||||
| QC-2(100%) | 88.36 | 87.55 87.76 87.34 |
99.1 99.3 98.9 |
99.1 | ||||||
| QC-3(120%) | 106.0 | 103.0 102.7 101.8 |
97.1 96.9 96.0 |
96.7 | ||||||
| Total average(%) | 98.2 | |||||||||
| Total RSD(%) | 1.2 | |||||||||
Abbreviation : QC mean Quality Control Sample, RSD mean Relative Standard Deviation, aFound conc.(ug/mL) : (QC sample peak area ? intercept) / slope, bAccuracy : Found conc. / Spiked conc. x 100, cPrecision(RSD) : SD(Found concs,) / Average(Found concs.) x 100, *Calculated from the summation of rosuvastatin and its degradant peaks
Table 5: Results of Accuracy And Precision Test for Telmisartan (Medium : Ph1.2, Ph4.0, Ph6.8, Water)
Estimates of the limit of quantitation based on the SD of the y-intercept and the slope of the calibration curve were 0.128-0.582 µg/ml for rosuvastatin calcium and 0.232-1.200 µg/ml for telmisartan, which corresponded to the lower level of the calibration curve.
As shown in Table 6, the limits of quantitation for rosuvastatin calcium based on the lowest concentration yielding acceptable accuracy and precision were 0.128 µg/ml at pH 1.2, 0.531 µg/ml at pH 4.0, 0.582 µg/ml at pH 6.8 and 0.472 µg/ml in water. The limits of quantitation for telmisartan based on the lowest concentration yielding acceptable accuracy and precision were 0.232 µg/ml at pH 1.2, 1.20 µg/ml at pH 4.0, 0.677 µg/ml at pH 6.8 and 0.447 µg/ml in water.
| medium | pH1.2 | pH4.0 | pH6.8 | Water | |||||
|---|---|---|---|---|---|---|---|---|---|
| LOQ(ug/mL) | Rosuvastatin calcium | 0.128 | 0.531 | 0.582 | 0.472 | ||||
| Telmisartan | 0.232 | 1.200 | 0.677 | 0.447 | |||||
| LOD(ug/mL) | Rosuvastatin calcium | 0.042 | 0.175 | 0.192 | 0.156 | ||||
| Telmisartan | 0.076 | 0.396 | 0.223 | 0.147 | |||||
Abbreviation : LOQ means limit of quantitation, LOD means limit of detection
Table 6: Limit of Quantitation and Limit of Detection of Rosuvastatin Calcium and Telmisartan in Various Dissolution Media
Estimates of the limit of detection based on the SD of the y-intercept and the slope of the calibration curve were 0.042-0.192 µg/ml for rosuvastatin calcium and 0.076-0.396 µg/ml for telmisartan. As shown in Table 6, the limits of detection for rosuvastatin calcium based on detectable concentrations were 0.042 µg/ml at pH 1.2, 0.175 µg/ml at pH 4.0, 0.192 µg/ml at pH 6.8 and 0.156 µg/ml in water. The limits of detection for telmisartan based on detectable concentrations were 0.076 µg/ml at pH 1.2, 0.396 µg/ml at pH 4.0, 0.223 µg/ml at pH 6.8, and 0.147 µg/ml in water.
For simultaneous quantitative analysis, it is important to demonstrate the robustness of the method to ensure that the HPLC method is not affected by minor changes in the experimental conditions. In all experiments, none of the alterations caused a significant change in resolution between rosuvastatin calcium and telmisartan peak area RSD, USP tailing factor, peak width, or theoretical plates. Because the robustness was sufficiently identified through the change in pump flow rate (±10 %), auto-sampler injector volume (±50 %) and column compartment temperature (±4°), the robustness can be considered acceptable. In addition, the robustness might be sufficiently secured because the test was conducted according to the FDA method validation regulations.
As shown in Table 7, when the solution stability between the standard samples and references samples for rosuvastatin and telmisartan in the four dissolution test solutions were compared, rosuvastatin calcium was stable to within 3.0 %, with no significant differences at 25° over 26 h at the pH 1.2, 27 h at pH 4.0, 30 h at pH 6.8 and 30 h in water. Telmisartan was also stable to within 3.0%, with no significant differences at 25° over 26 h at the pH 1.2, 50 h at pH 4.0, 30 h at pH 6.8, and 30 h in water. It was confirmed that both rosuvastatin calcium and telmisartan displayed no significant difference within 5 %, which is a reasonably acceptable criterion for all dissolution media at 25° over the course of 30 h.
| Solution stability | Medium | Time | Standard conc.(ug/mL) | Difference(%) | Time | Sample conc.(ug/mL) | Difference(%) |
|---|---|---|---|---|---|---|---|
| Rosuvastatin calcium | pH1.2 | Initial 23hr 26hr |
22.35 22.33 22.36 |
- -0.15 -0.38 |
Initial 23hr 26hr |
22.14 22.23 22.23 |
- -0.44 -0.44 |
| pH4.0 | Initial 24hr 27hr |
22.37 22.36 22.38 |
- 0.62 0.56 |
Initial 24hr 27hr |
22.17 21.57 22.02 |
- 2.71 0.67 |
|
| pH6.8 | Initial 28hr 30hr |
11.22 11.19 11.20 |
- -1.23 -0.77 |
Initial 28hr 30hr |
11.26 11.11 11.25 |
- 1.37 0.14 |
|
| Water | Initial 28hr 30hr |
11.22 11.19 11.20 |
- 0.82 -1.22 |
Initial 28hr 30hr |
11.01 11.15 11.28 |
- -1.25 -2.41 |
|
| Telmisartan | pH1.2 | Initial 23hr 26hr |
88.40 88.39 88.46 |
- -0.84 0.46 |
Initial 23hr 26hr |
89.92 89.23 88.88 |
- 0.77 1.16 |
| pH4.0 | Initial 25hr 50hr |
17.78 17.76 17.77 |
- -2.84 0.44 |
Initial 25hr 50hr |
18.19 18.29 17.71 |
- -0.56 2.61 |
|
| pH6.8 | Initial 28hr 30hr |
44.18 44.16 44.24 |
- -1.83 -0.61 |
Initial 28hr 30hr |
43.05 43.08 43.44 |
- -0.09 -0.92 |
|
| Water | Initial 28hr 30hr |
44.18 44.16 44.24 |
- 0.30 0.67 |
Initial 28hr 30hr |
43.75 42.59 44.17 |
- 2.65 -0.97 |
Table 7: Results of Solution Stability for Rosuvastatin Calcium and Telmisartan in Various Dissolution Media
We compared the dissolution profiles of our developed tablets and commercial products. In the case of telmisartan, the similarity factors in each dissolution test solution were identified as 58.72, 62.41, 64.58 and 65.93 at pH 1.2, pH 4.0, pH 6.8, and in water, respectively. In the case of rosuvastatin calcium, the similarity factors in each dissolution test solution were identified as 63.47, 66.81, 59.68 and 60.12 at pH 1.2, pH 4.0, pH 6.8 and in water respectively. Therefore, the developed telmisartan and rosuvastatin calcium FDC dual-layer tablets and commercial products exhibit similar dissolution profiles in the entire pH range. Generally, pH 1.2, 4.0, 6.8 and water are used as dissolution test solutions to predict the pharmacokinetics of drugs. The pH 1.2 solution simulates gastric juice, the pH 4.0 solution simulates the duodenum environment and the pH 6.8 solution simulates the colon environment. In addition, the FDA’s “Dissolution Testing of Immediate Release Solid Oral Dosage Forms” states that dissolution tests should be conducted within the range of pH 1.2-6.8. In addition, the result at pH 4.0 could replace the dissolution test at pH 4.5. Thus, we have predicted the in vitro dissolution profiles of the tablet through the dissolution test at pH 4.0.
The validated method was used in the analysis of six rosuvastatin calcium and telmisartan drug products (telmisartan and rosuvastatin calcium FDC dual-layer tablets with dosages of 80/20, 80/5, 80/10, 40/20, 40/5 and 40/10 mg). Rosuvastatin calcium and telmisartan FDC dual-layer tablets were analyzed through simultaneous quantitative analysis in the dissolution samples. As a result of the method validation in the dissolution sample, each performance characteristic satisfied the recommended guidelines. We confirmed suitability using simultaneous quantitative analysis with rosuvastatin calcium concentrations in the range of 4.44-26.6 µg/ml and with telmisartan concentrations in the range of 0.176-106.6 µg/ml for pH 1.2, 4.0, 6.8 and water dissolution samples. The simultaneous quantitative analysis described in this work could reduce cost and time and could be a valuable tool for the pharmaceutical industry.
Acknowledgements:
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (NRF-2018R1C1B5045232).
References
- Quirk J, Thornton M, Kirkpatrick P. Rosuvastatin calcium. Nat Rev Drug Discov 2003;2(10):769.
- Olsson AG, McTaggart F, Raza A. Rosuvastatin: a highly effective new HMG-CoA reductase inhibitor. Cardiovasc Drug Rev 2002;20(4):303-28.
- Pasha MK, Muzeeb S, Basha SJ, Shashikumar D, Mullangi R, Srinivas NR. Analysis of five HMG-CoA reductase inhibitors-atorvastatin, lovastatin, pravastatin, rosuvastatin and simvastatin: pharmacological, pharmacokinetic and analytical overview and development of a new method for use in pharmaceutical formulations analysis and in vitro metabolism studies. Biomed Chromatogr 2006;20(3):282-93.
- Mc Clellan KJ, Markham A. Telmisartan. Drugs 1998;56(6):1039-44.
- Potale LV, Damle MC, Khodke AS, Bothara KG. A validated stability indicating HPTLC method for simultaneous estimation of ramipril and telmisartan. Int J Pharm Sci Rev Res. 2010;2:7.
- Patil KR, Shinde DB. Stability-indicating LC method for the simultaneous determination of Telmisartan and Hydrochlorothiazide in dosage form. J Chile Chem Soc 2012;57(1):1017-21.
- Kang WY, Kim W, Hwang SH, Hong KH, Rho HM, Park CS, et al. Effects of telmisartan and rosuvastatin for endothelial function and arterial stiffness in hypertensive patients in Korea. Int J Cardiol 2009;137:S35-6.
- Chen Y, Liu H, Wang J, Zhou T, Sun S, Zhang H, et al. Effect of telmisartan combined with rosuvastatin on white matter lesion in elderly hypertensive patients. Chin J Geriatr Heart Brain Vessel Dis 2018;20(2):145-8.
- Hong SJ, Jeong HS, Cho JM, Chang K, Pyun WB, Ahn Y, et al. Efficacy and safety of triple therapy with telmisartan, amlodipine, and rosuvastatin in patients with dyslipidemia and hypertension: The jeil telmisartan, amlodipine, and rosuvastatin randomized clinical trial. Clin Ther 2019;41(2):233-48.
- Shah Y, Iqbal Z, Ahmad L, Khan A, Khan MI, Nazir S, et al. Simultaneous determination of rosuvastatin and atorvastatin in human serum using RP-HPLC/UV detection: Method development, validation and optimization of various experimental parameters. J Chromatogr 2011;879(9-10):557-63.
- Patel VA, Patel PG, Chaudhary BG, Rajgor NB, Rathi SG. Development and validation of hptlc method for the simultaneous estimation of telmisartan and ramipril in combined dosage form. Int J Pharm Biol Res 2010;1(1):18-24.
- Palled MS, Rajesh PN, Chatter M, Bhat AR. RP-HPLC determination of Telmisartan in tablet dosage forms. Ind J Pharm Sci 2005;67(1):108.
- Kaila HO, Ambasana MA, Thakkar RS, Saravaia HT, Shah AK. A new improved RP-HPLC method for assay of rosuvastatin calcium in tablets. Ind J Pharm Sci 2010;72(5):592.
- Shinde NG, Aloorkar NH. Development and validation of UV spectrophotometric method for simultaneous estimation of propranolol hydrochloride and rosuvastatin calcium in bulk drug and pharmaceutical dosage form. Int J Adv Pharm 2015;4(5):1-5.
- Sangeetha D, Vadlamudi MK. Development and Validation of a Stability-indicating RP-HPLC Method for Estimation of Metformin and Rosuvastatin along with Impurities from a Combined Oral Solid Dosage Form. Ind J Pharm Sci 2019;81(2):365-74.
- Peddapalli H. Method Development and Validation for the Simultaneous Estimation of Rosuvastatin and Amlodipine in Bulk and its Formulation using Reverse-Phase High-Performance Liquid Chromatography. Asian J Pharm 2019;12(4):8.
- Palur K, Koganti B, Archakam SC. Chemometric-assisted RP-HPLC method for the simultaneous determination of ambroxol hydrochloride, terbutaline sulfate, and guaiphenesin in combined dosage form. J Applied Pharm Sci 2019;9(09):92-7.
- Doshi N, Sheth A, Patel T, Dave JB, Patel CN. Spectrophotometric absorption factor method development and validation for estimation of Rosuvastatin Calcium and Telmisartan in solid dosage form. J Chem Pharm Res 2010;2(3):15-24.
- Pandya CB. Development and validation of RP-HPLC method for determination of rosuvastatin calcium in bulk and pharmaceutical dosage form. Int J Pharm Sci Rev Res 2010;5:82-6.
- Epshtein NA. Validation of HPLC techniques for pharmaceutical analysis. Pharm Chem J 2004;38(4):212-28.
- Taverniers I, Van Bockstaele E, De Loose M. Analytical method validation and quality assurance. Pharm Sci Encyclopedia Drug Discov Develop 2010;15:1-48.
- Ermer J, Miller JH, editors. Method validation in pharmaceutical analysis: A guide to best practice. John Wiley & Sons; 2006.
- Bhat LR, Godge RK, Vora AT, Damle MC. Validated Rp-HPLC method for simultaneous determination of Telmisartan and Hydrochlorothiazide in pharmaceutical formulation. J Liquid Chromatogr 2007;30(20):3059-67.
- Venkatasami G, Sowa Jr JR. A rapid, acetonitrile-free, HPLC method for determination of melamine in infant formula. Analytica Chimica Acta 2010;665(2):227-30.
- Carr GP, Wahlich JC. A practical approach to method validation in pharmaceutical analysis. J Pharm Biomed Anal 1990;8(8):613-8.
- Nasir F, Iqbal Z, Khan A, Ahmad L, Shah Y, Khan AZ, et al. Simultaneous determination of timolol maleate, rosuvastatin calcium and diclofenac sodium in pharmaceuticals and physiological fluids using HPLC-UV. J Chromatogr 2011;879(30):3434-43.
- Wiggins DE. System suitability in an optimized HPLC system. J Liquid Chromatogr 1991;14(16-17):3045-60.
- Huber L. Validation of HPLC methods. BioPharm 1999;12(3):64-6.
- Vander Heyden Y, Nijhuis A, Smeyers-Verbeke J, Vandeginste BG, Massart DL. Guidance for robustness/ruggedness tests in method validation. J Pharm Biomed Anal 2001;24(5):723-53.
- Subramanian VB, Katari NK, Dongala T, Jonnalagadda SB. Stability-indicating RP-HPLC method development and validation for determination of nine impurities in apixaban tablet dosage forms. Robustness study by quality by design approach. Biomed Chromatogr 2020;34(1):e4719.