- *Corresponding Author:
- Jingyi Zhao
Key Laboratory of Traditional Chinese Medicine Research and Development of Hebei Province, Institute of Traditional Chinese Medicine, Chengde Medical University, China
E-mail: 33236986@qq.com
| This article was originally published in a special issue, “New Research Outcomes in Drug and Health Sciences” |
| Indian J Pharm Sci 2023:85(6) Spl Issue “141-151” |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
By using network pharmacology, molecular docking and molecular dynamics, active ingredients, targets and mechanisms of Erigeron breviscapus in treating myocardial ischaemia-reperfusion injury were elucidated. It was found that the target gene for Erigeron breviscapus crossed with the disease gene related to myocardial ischaemia-reperfusion injury after obtaining the drug components. According to those intersection results, cytoscape was used to obtain the hub genes. The hub genes were subjected to gene ontology enrichment and analyzed with the Kyoto Encyclopedia of Genes and Genomes. After molecular docking of the hub genes with the components of Erigeron breviscapus, the optimal docking result was selected for molecular dynamics simulation. The two main active ingredients (1-hydroxy-2,3,5-trimethoxyxanthone and scutevulin) of Erigeron breviscapus have 51 therapeutic targets related to myocardial ischaemia-reperfusion injury, including 6 key genes (epidermal growth factor receptor, estrogen receptor 1, matrix metalloproteinase, prostaglandin-endoperoxide synthase 2, heat shock protein 90 alpha family class A member 1 and serine/ threonine kinase 1). The signaling pathways mainly focus on inflammatory reactions. The results of molecular docking and molecular dynamics show that scutevulin closely and stably binds with estrogen receptor 1 and with prostaglandin-endoperoxide synthase 2. In the treatment of myocardial ischaemia-reperfusion injury, Erigeron breviscapus may act on target genes such as estrogen receptor and prostaglandin-endoperoxide synthase 2 that affect the signal pathway of inflammatory factors.
Keywords
Erigeron breviscapus, myocardial ischaemia-reperfusion injury, network pharmacology, molecular docking, molecular dynamics simulation
In Myocardial Ischaemia-Reperfusion Injury (MIRI), the myocardial blood supply is interrupted for a short time and then restored to the ischaemic myocardium within a certain period of time, however, the resulting injury is more serious than that before blood supply recovery[1,2]. Clinically, MIRI occurs when an occluded coronary artery is recanalized. In some cases, blood pressure drops, cardiac insufficiency, arrhythmia and other problems, even sudden death can occur[3-6]. The effects of ischemia worsen after a period of blood perfusion restoration to the infarcted area. Studies on the mechanism of reperfusion injury have shown that oxygen free radicals[7], calcium overload[8], energy metabolism disorder of myocardial fibres[9], vascular endothelial cells[10], nitric oxide[11], neutrophils and apoptosis may all be involved in the pathogenesis of MIRI[12]. Prevention and mitigation of MIRI are key to surgical treatment of ischaemic heart disease and early reperfusion of thrombolysis or primary percutaneous coronary intervention is a common measure for clinical treatment of MIRI[13]. Therefore, it is necessary to study safer and more effective treatment interventions to reduce the clinical risk of MIRI and provide new treatment strategies.
Traditional Chinese Medicine (TCM) as an important field of global medicine, plays an irreplaceable role in the treatment of MIRI. Erigeron breviscapus, also known as Chrysanthemum morifolium, is abundant in Yunnan[14]. The medicine is the dry whole grass of Erigeron breviscapus, which is a composite plant. Recent studies have shown that Erigeron breviscapus has many pharmacological effects including-improves blood circulation[15], anti-inflammatory[16] and antioxidative effects[17]. Network pharmacology is a promising method of TCM evaluation. Systems biology can be combined with pharmacokinetics and pharmacodynamics to study drugs, targets and their pharmacological activities. Therefore, in this study, computational tools and resources were used to study the pharmacological network of the effects of Erigeron breviscapus on MIRI to predict the active compounds and potential protein targets. Molecular docking and Molecular Dynamics (MD) assessments were used to detect the interactions between activated compounds and protein targets.
Materials and Methods
Active component screening and targets:
High-throughput Experiment-and Reference-guided database of TCM (HERB) (http://herb.ac.cn/) is an efficient network pharmacology resource. Its database contains 1241 gene targets and 494 modern diseases affected by herbs (or ingredients)[18]. In this study, the key words Erigeron breviscapus were entered into HERB to find the ingredients. To further understand the Absorption, Distribution, Metabolism and Excretion (ADME) characteristics of Erigeron breviscapus, the components of Erigeron breviscapus were predicted in Absorption, Distribution, Metabolism, Excretion and Toxicity lab (ADMETlab) 2.0 (https://admetmesh.scbdd.com/)[19]. The main indexes of ADME examined in this study were oral bioavailability (F %) (20 %)≥0-0.3 and Quantitative Estimate of Drug-likeness (QED)>0.67. F % is a reliable index to objectively evaluate the internal qualities of drugs that represents the speed and degree of drug absorption into the circulatory system. Drug-likeness represents the sum of the pharmacokinetic properties and safety of compounds, which is calculated by comparing the functional or physical properties of compounds with those of most known drugs. The chemical composition of Erigeron breviscapus found in the HERB database was submitted to the SwissTargetPrediction (http://www.swisstargetprediction.ch/) tool to predict the targets of the components[20].
Mining of MIRI-related targets:
Targets related to MIRI were collected through the Online Mendelian Inheritance in Man (OMIM) database (https://www.omim.org/) and GeneCards database (https://www.genecards.org/). The targets of both the databases were uploaded to the Universal Protein resource (UniProt) database (http://www.uniprot.org/) and the species was defined as “Homo sapiens” to obtain the corresponding gene information and correct the targets. Then, the targets corresponding to the compounds of Erigeron breviscapus and the targets related to MIRI were intersected to obtain the potential targets of the drug micro molecules.
Construction of herb-active ingredient-target interaction network:
The network of active ingredient targets of the TCM, Erigeron breviscapus was constructed by using Cytoscape 3.9.0 (http://cytoscape.org/), which can help visually display the pharmacological mechanisms[21].
Construction and analysis of a Protein-Protein Interaction (PPI) network:
The potential targets of the compounds in Erigeron breviscapus were introduced into the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) 11.0 database (https://string-db.org)[22], and the species was defined as “Homo sapiens” to construct the PPI network model. The confidence of the network was>0.4 and the unconnected nodes were hidden. The other parameters were set to the default values. Finally, a PPI network was obtained and the constructed network was downloaded in parallel Tab-Separated Values (TSV) file format and then imported into Cytoscape 3.9.0 software to visualize the PPI network. We used the Cytoscape app for Network Centrality Analysis (CytoNCA) plug-in tool in Cytoscape to analyze the protein targets in the complex bioinformatics unweighted network[23]. The analysis methods included the Degree Centrality (DC), Betweenness Centrality (BC), Closeness Centrality (CC), Eigenvector Centrality (EC), Information Centrality (IC), Local Average Connectivity (LAC), Subgraph Centrality (SC) and Normalized alpha (α)-Centrality (NC) methods. Target screening was based on the inclusion criteria of median values greater than the topological parameters and the nodes ultimately obtained were considered as the key nodes in this interactive network.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment:
With R packages such as org.Hs.eg.db and clusterProfiler[24], with p<0.05 as the screening condition, GO enrichment analysis and KEGG signaling pathway analysis of the target functions in key nodes were carried out.
Molecular docking:
The crystal structures of the gene targets were obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) (https://www.rcsb.org/). The crystal water and complex small molecules were removed by Proprietary Molecular visualization system (PyMOL) 2.4.1 software and then imported into AutoDock tools 1.5.6 software for hydrogenation and charging. The result was output in Protein Data Bank, Partial Charge and Atom Type (PDBQT) format. Small drug molecules were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) and imported into ChemBio3D Ultra 14.0 software and their energy was minimized. The Cavity-detection guided Blind Docking (CB-Dock) online tool was used for molecular docking (http://cao.labshare.cn/cbdock//)[25]. The Vina fraction, a lower value of that indicates more stable binding of ligand to a receptor, which was used preliminarily to evaluate the compound-target binding activity. The complex was imported into Discovery Studio 2016 software and the distance of hydrogen bond and the number of pi bonds were calculated.
MD simulation:
The initial structures of the target proteins were obtained from the RCSB PDB. Through the Preparing PDB structure (Advanced Edition) scheme of Yin Fuyun's computing platform (https://cloud.yinfotek.com/), the LEaP modules of PDBFixer and AmberTools 20[26,27] were called to prepare the receptors and repair the missing residue atoms. Based on the electrostatic potential calculated by Gaussian 09W[28] at the Becke, 3-parameter, Lee- Yang-Parr (B3LYP)/6-311G(d,p) level, the semi- empirical with Bond Charge Correction (AM1-BCC) charges of small molecules were calculated by the Amber antechamber program[29].
On the Yin Fuyun computing platform, AmberTools 20 was called to carry out MD simulation and the Assisted Model Building with Energy Refinement force field 2019 Stony Brook (AMBER ff19SB) [30], General Amber Force Field (GAFF)[31] were used for proteins and small molecules, respectively. The Optimal Point Charge (OPC) water model and truncated octahedral water box with edge 10 were used for solvent treatment. Periodic Boundary Conditions (PBCs) were used and the net charge was neutralized with 0.15 mol Sodium chloride (NaCl). Nonbonding van der Waals interactions were calculated by the L-J 12-6 potential and the truncation value was 10 Å, while the long-range electrostatic force was treated with the Principal Manifold Estimation (PME)[32] algorithm. The SHAKE algorithm was used to constrain covalent bonds involving hydrogen atoms[33]. To eliminate unfavorable interatomic contact, first a binding force of 10.0 kcal/(mol Å2) was applied to the heavy atoms and the energy was minimized by the 2500-step steepest descent method and 2500-step conjugate gradient method. Then, the whole system was relaxed and the energy was minimized by the 10 000-step steepest descent method and 10 000-step conjugate gradient method. The temperature of the system was gradually raised to 300 K by 20 picosecond (ps) constant-Number (N), constant-Volume (V), and constant-Temperature (T) (NVT) simulation, after which the two-step equilibrium phase was carried out. First, 200 ps of constrained heavy atom constant- Temperature, constant-Pressure ensemble (NPT) simulation was performed; next, 1 nanosecond (ns) of unconstrained NVT simulation was performed. A Berendsen hot bath was used to control the temperature at 300 K, a Monte Carlo pressure bath was used to control the pressure at 1 atm and the coupling constant and relaxation time were both 1 ps. Finally, a 20 ns NVT simulation was carried out and the integration time step was 2 femtosecond (fs) [34]. According to the following formula, a total of 200 snapshots were extracted from MD trajectories and used to calculate the binding free energy (ΔGbind) of Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA)[35]
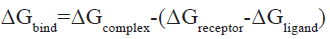
Results and Discussion
Active ingredients and target genes of Erigeron breviscapus were found. A total of 14 active ingredients of Erigeron breviscapus were found in the HERB database. Fourteen small molecules were predicted by ADME in ADMETlab 2.0 and two small molecules (1-hydroxy-2,3,5-trimethoxyxanthone and scutevulin) were finally screened according to F % (20 %)≥0-0.3 and QED>0.67. The targets of 1-hydroxy-2,3,5-trimethoxyxanthone and scutevulin were predicted by the SwissTargetPrediction database.1-Hydroxy-2,3,5-trimethoxyxanthone had 81 target genes, and scutevulin had 100 target genes. A total of 169 target genes were obtained by removing duplicate genes from the targets of the two compounds. Through the OMIM database and GeneCards database, 1153 targets related to MIRI were retrieved and 51 common target genes were obtained by crossing the potential target genes of 1-hydroxy-2,3,5-trimethoxyxanthone and scutevulin (fig. 1).

Fig. 1: Venn plot of 51 potential targets
Further, compound-target network was predicted for which the protein interaction data of 51 targets was obtained by the STRING online analysis tool and were imported into Cytoscape software. The TCM- active ingredient-target network including Erigeron breviscapus and its active ingredients is displayed, which contains 54 nodes and 298 edges in this network (fig. 2).

Fig. 2: Erigeron breviscapus-MIRI-potential target gene network
The analysis of key targets of Erigeron breviscapus was carried out. For this, the PPI network diagram of 51 targets was analyzed with the CytoNCA tool of Cytoscape software. The median values of BC, CC, DC, EC, IC, LAC, NC and SC were 55.37, 0.48, 9.41, 0.11, 3.69, 4.39, 6.73 and 52547.14, respectively. Then, 16 key targets were obtained and a network diagram was constructed. When the same analysis method mentioned above was used, the median values of BC, CC, DC, EC, IC, LAC, NC and SC were 4.5, 0.78, 10.5, 0.24, 5.76, 7.40, 9.46 and 4513.65 respectively, and ultimately, six key targets Epidermal Growth Factor Receptor (EGFR), Estrogen Receptor 1 (ESR1), Matrix Metalloproteinase (MMP9), Prostaglandin-Endoperoxide Synthase 2 (PTGS2), Heat Shock Protein 90 Alpha Family Class A Member 1 (HSP90AA1) and Serine/Threonine Kinase 1 (AKT1) were obtained (fig. 3).

Fig. 3: Topological analysis of the intersecting targets between MIRI-related targets and targets of Erigeron breviscapus active compounds
GO and KEGG pathway enrichment analysis was evaluated in which GO enrichment analysis of the above six key targets showed that the main enriched Biological Process (BP) terms were the response to Ultraviolet A (UV-A), positive regulation of smooth muscle cell proliferation and positive regulation of peptidyl-serine phosphorylation. The Molecular Function (MF) terms were Adenosine Triphosphatase (ATPase) binding, protein phosphatase binding and phosphatase binding. The Cellular Component (CC) terms were ficolin-1-rich granule lumen, ficolin-1- rich granule and transcription preinitiation complex (fig. 4). KEGG enrichment analysis revealed that these genes were significantly enriched in the Interleukin (IL)-17 signaling pathway, Tumor Necrosis Factor (TNF) signaling pathway, relaxin signaling pathway, Phosphoinositide-3-Kinase-Protein Kinase B (PI3K- Akt) signaling pathway and Janus Kinase-Signal Transducers and Activators of Transcription (JAK- STAT) signaling pathway (fig. 4).

Fig. 4: GO enrichment and KEGG pathway enrichment analysis
Note:  KEGG
KEGG
Further, for molecular docking two active ingredients (1-hydroxy-2,3,5-trimethoxyxanthone and scutevulin) were predicted to bind with six key targets at different degrees (Table 1 and fig. 5). A lower Vina score indicated that an interaction between compounds and receptors was stronger and more stable. Scutevulin had the strongest and most stable binding affinity for ESR1 and PTGS2.
| Molecule name | Target name | PDB ID | Score (kcal/mol) | Cavity size | Centre | Size | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| x | y | z | x | y | z | |||||
| 1-Hydroxy-2,3,5-trimethoxyxanthone | PTGS2 | 5F19 | -8.4 | 1247 | 31.6 | 37.6 | 17.8 | 20 | 20 | 33 |
| HSP90AA1 | 1BYQ | -7 | 705 | 40.5 | -48 | 64.6 | 20 | 20 | 20 | |
| Scutevulin | PTGS2 | 5F19 | -9.1 | 5179 | 11.9 | 52.6 | 16.7 | 33 | 26 | 29 |
| MMP9 | 1GKD | -7.5 | 940 | -2.4 | 22.3 | 16.7 | 20 | 31 | 20 | |
| HSP90AA1 | 1BYQ | -7.4 | 705 | 40.5 | -48 | 64.6 | 20 | 20 | 20 | |
| ESR1 | 1GWR | -9 | 1719 | 9.96 | 2.37 | -16.7 | 32 | 20 | 20 | |
| EGFR | 1M14 | -7.7 | 3283 | 29.1 | 9.37 | 50.3 | 32 | 35 | 20 | |
| AKT1 | 1UNP | -6.1 | 206 | 24.9 | -4.4 | 15.9 | 20 | 20 | 20 | |
Table 1: Molecular Docking Results of Targets and their Active Components

Fig. 5: Comprehensive analysis of Erigeron breviscapus active compound-key target network with molecular docking
Note:  pi-pi T-shaped bond;
pi-pi T-shaped bond;  pi-Alkyl bond
pi-Alkyl bond
As PTGS2 and ESR1 were the selected core targets, the binding of scutevulin with these two targets in the molecular docking assessment was superior. Therefore, we chose the docking conformations of PTGS2, scutevulin, ESR1 and scutevulin with low binding energy as the initial conformations for the MD simulation (fig. 6). The Root Mean Square Deviation (RMSD), which is an important basis to measure the stability of a system was calculated as the sum of all atomic deviations between the conformation and the target conformation at a certain moment. Low RMSD values of PTGS2 (0.57-1.29), scutevulin (0.78-1.75) and ESR1 (0.6-1.33) are shown in fig. 6. The results indicate that the molecular docking of scutevulin with PTGS2 or ESR1 is very stable. The MD trajectory was calculated by Molecular Mechanics with Generalised Born and Surface Area Solvation (MMGBSA) to study the binding energy of the small molecules and targets. Table 2 summarizes the results of the two complexes in the MD simulation. The results show that all binding free energies are less than zero, which indicates that the reaction can proceed spontaneously.
| Complexes | ΔGvdw | ΔGele | ΔGpolar | ΔGnonpolar | ΔGgas | ΔGsolv | ΔGtotal |
|---|---|---|---|---|---|---|---|
| Scutevulin-PTGS2 | -37.10±2.43 | -11.82±3.54 | 26.05±2.28 | -3.57±0.081 | -48.93±3.24 | 22.48±2.29 | -26.45±2.95 |
| Scutevulin-ESR1 | -37.95±2.37 | -13.12±2.40 | 32.75±3.34 | -3.60±0.074 | -51.07±2.88 | 29.14±3.34 | -21.93±3.47 |
Note: All energies are in kcal mol-1 with the standard deviation in parentheses (ΔGvdw): van der Waals energy; (ΔGele): Electrostatic energy; (ΔGpolar): Polar solvation energy; (ΔGnonpolar): Nonpolar solvation energy; (ΔGgas): Energy in gas phase; (ΔGsolv): Solvation energy; (ΔGtotal): Total binding free energy
Table 2: Binding Free Energy Values of Each Complex and Various Energy Components
In recent years, the incidence of Coronary Atherosclerotic Heart Disease (CAHD) has increased year by year and the increased incidence is accompanied by an increase in mortality. Interventional therapy, one of the main methods used to treat CAHD can be associated with MIRI[36,37]. The occurrence of MIRI usually leads to poor prognosis and can even endanger the lives of patients. Therefore, safe and effective ways to prevent MIRI are urgently needed. Many network pharmacology-based studies have been performed to investigate the mechanisms of action of Erigeron breviscapus on cardiovascular diseases.

Fig. 6: RMSD of the composite system
Note:  Protein RMSD
Protein RMSD
Previous studies have also revealed that scutellarin, a component of Erigeron breviscapus, which can protect against MIRI through its ingredient baicalein. Certain proteins may regulate oxidative stress and apoptosis induced by MIRI by enhancing Janus Kinase 2/Signal Transducer and Activator of Transcription 3 (JAK2/STAT3) prosurvival signaling[38]. Scutellarin also provides cardioprotection through its anti-inflammatory effects. For example, it inhibits activation of the Nucleotide-binding domain, Leucine- Rich-containing family, Pyrin domain-containing-3 (NLRP3) inflammasome. Scutellarin's restriction of inflammatory bodies depends on activation of Akt and inhibition of mammalian Target of Rapamycin Complex 1 (mTORC1)[39]. In addition, the protective effect of scutellarin against hypoxia-reoxygenation injury of human cardiac microvascular endothelial cells is related to Eukaryotic translation Initiation Factor 6 (EIF6), Heat Shock Protein Family D 1 (HSPD1) and Chaperonin-Containing T-Complex Protein 1 (TCP1) Subunit 6A (CCT6A)[40]. Wang et al. reported that Erigeron breviscapus can alleviate cardiac fibrosis, regulate myocardial contractility and protect cardiac function when cardiac remodelling is poor. It can also promote blood circulation and dilate blood vessels[41].
In this study, a variety of chemical components of Erigeron breviscapus were first screened, and after F % and drug-likeness screening, 1-hydroxy-2,3,5-trimethoxyxanthone and scutevulin were identified as the research focuses. Protein interactions play important roles in key biological processes. In this study, the CytoNCA plug-in tool was used and identified six potential key targets (EGFR, ESR1, MMP9, PTGS2, HSP90AA1 and AKT1) of 1-hydroxy-2,3,5-trimethoxyxanthone and scutevulin in the treatment of MIRI. The signaling pathways (IL-17 signaling pathway, TNF signaling pathway, relaxin signaling pathway, PI3K/Akt signaling pathway and JAK-STAT signaling pathway) associated with these six targets and MIRI have been reported. IL-17 plays a pathogenic role in MIRI, so inhibition of IL-17 can protect myocardial cells[42]. The reduction in cardiac ischaemia-reperfusion injury mediated by necrostin-1 is closely related to inhibition of the High-Mobility Group Box 1 Protein- Interleukin-23/Interleukin-17 (HMGB1-IL-23/IL-17) pathway[43]. Therefore, the protective effect of Erigeron breviscapus against MIRI may originate from IL-17 signaling pathways related to MMP9, PTGS2 and HSP90AA1. Nuclear Factor-Kappa B (NF-κB), a key factor regulating gene transcription, participates in the regulation of early defense and the inflammatory response[44,45]. Previous studies have shown that the NF-κB pathway plays a biological role in the production of Reactive Oxygen Species (ROS) and the infiltration of polymorphonuclear neutrophils during MIRI[46,47]. Relaxin is a pleiotropic and cardioprotective hormone that has been indicated to promote vasodilation and angiogenesis, improve ischaemia-reperfusion injury and regulate extracellular matrix renewal and remodelling[48,49]. Thus far, however, there has been little research on the effects of drugs on the relaxin signaling pathway. Our findings will provide a new direction for research on the mechanism of Erigeron breviscapus in MIRI protection. The PI3K-Akt signaling pathway, one of the most important signaling pathways regulating cell survival, is closely related to myocardial protection in MIRI[50,51]. Therefore, the role of the PI3K-Akt signaling pathway in MIRI has attracted much attention. The JAK-STAT signaling pathway plays key regulatory roles in cell growth, development, proliferation, apoptosis and necrosis[52]. After MIRI, the JAK-STAT signaling pathway can be activated quickly[53]. In particular, phosphorylation of JAK2 and STAT3 is initiated to mediate cardiomyocyte apoptosis, which is also an important pathological mechanism of apoptosis after MIRI[54,55].
The results of molecular docking showed that the Vina fractions of scutevulin with PTGS2 and ESR1 were -9.1 and -9 kcal/mol, respectively, while the Vina fraction of 1-hydroxy-2,3,5-trimethoxyxanthone and PTGS2 was -8.4 kcal/mol. Generally, when the Vina value is <-7 kcal/mol, the intermolecular binding force is strong. 1-Hydroxy-2,3,5-trimethoxyxanthone and scutevulin, as the main active ingredients in Erigeron breviscapus, deserve further study in the context of MIRI treatment. Molecular docking further showed that the binding of scutevulin and PTGS2 and the binding of scutevulin and ESR1 were superior. The MD analysis also indicated that scutevulin might act on PTGS2 and ESR1 to treat MIRI. The results of the MD trajectory also showed that the binding of scutevulin with PTGS2 and the binding of scutevulin with ESR1 were most stable, with free energies of -26.45 kcal/mol and -21.93 kcal/mol, respectively. 1-Hydroxy-2,3,5-trimethoxyxanthone also bound closely with PTGS2. Molecular docking and MD simulation are computational methods that simulate the ability of compounds to bind to target proteins and cannot be used to draw conclusions regarding stimulation or inhibition. Therefore, the results of molecular docking and MD analysis should be carefully interpreted when 1-hydroxy-2,3,5- trimethoxyxanthone and scutevulin act on the same target, PTGS2.
This study was based on network pharmacology, molecular docking and MD simulation, which were used to perform a systematic analysis of the effects of Erigeron breviscapus in the treatment of MIRI. However, it should be noted that this study had some limitations. First, only potential targets of the components of Erigeron breviscapus have been detected thus far and these targets have not been proven to be related to MIRI. Second, verification of the results is needed to clarify the molecular mechanism of Erigeron breviscapus in MIRI treatment.
In summary, this study shows that 1-hydroxy-2,3,5- trimethoxyxanthone and scutevulin may be the key active ingredients of Erigeron breviscapus in MIRI treatment. The potential targets corresponding to the main components include EGFR, ESR1, MMP9, PTGS2, HSP90AA1 and AKT1. In addition, the effect of Erigeron breviscapus on MIRI may be achieved via regulation of the IL-17 signaling pathway. The molecular docking and MD simulation results show that both 1-hydroxy-2,3,5-trimethoxyxanthone and scutevulin have good affinity for the potential target PTGS2, which is worthy of further research in the future.
Funding:
We thank the Natural Science Foundation of Chengde Medical University (Grant no. KY2020015), Research on Science and Technology of Hebei Institution of Higher Learning in 2022 (Grant no. QN2022032), 2021 Research Start-up Fund for High-level Talents of Chengde Medical University (202109) and the Department of Neurobiology of Chengde Medical University (071006).
Conflict of interests:
The authors declared no conflict of interest.
References
- Inoue T. Ischemia-reperfusion injury is still a big hurdle to overcome for treatment of acute myocardial infarction. J Cardiol 2016;67(4):305-6.
[Crossref] [Google Scholar] [PubMed]
- Papageorgiou N, Briasoulis A, Tousoulis D. Ischemia-reperfusion injury: Complex pathophysiology with elusive treatment. Hellenic J Cardiol 2018;59(6):329-30.
[Crossref] [Google Scholar] [PubMed]
- Rios-Navarro C, Daghbouche-Rubio N, Gavara J, de Dios E, Perez N, Vila JM, et al. Ischemia-reperfusion injury to coronary arteries: Comprehensive microscopic study after reperfused myocardial infarction. Ann Anat 2021;238:1-10.
[Crossref] [Google Scholar] [PubMed]
- Wray S, Lascano E, Negroni J, Fischer EC. Relationship between augmentation index and wall thickening fraction during hypotension in an animal model of myocardial ischemia-reperfusion and heart failure. Curr Hypertens Rev 2021;17(2):121-30.
[Crossref] [Google Scholar] [PubMed]
- Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, et al. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol Biochem 2018;46(4):1650-67.
[Crossref] [Google Scholar] [PubMed]
- Lahnwong S, Palee S, Apaijai N, Sriwichaiin S, Kerdphoo S, Jaiwongkam T, et al. Acute dapagliflozin administration exerts cardioprotective effects in rats with cardiac ischemia/reperfusion injury. Cardiovasc Diabetol 2020;19(1):1-3.
[Crossref] [Google Scholar] [PubMed]
- Zhao WK, Zhou Y, Xu TT, Wu Q. Ferroptosis: Opportunities and challenges in myocardial ischemia-reperfusion injury. Oxid Med Cell Longev 2021;2021:1-12.
[Crossref] [Google Scholar] [PubMed]
- Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 2012;298:229-317.
[Crossref] [Google Scholar] [PubMed]
- Nuntaphum W, Pongkan W, Wongjaikam S, Thummasorn S, Tanajak P, Khamseekaew J, et al. Vagus nerve stimulation exerts cardioprotection against myocardial ischemia/reperfusion injury predominantly through its efferent vagal fibers. Basic Res Cardiol 2018;113(4):1-20.
[Crossref] [Google Scholar] [PubMed]
- Davidson SM, Ferdinandy P, Andreadou I, Bøtker HE, Heusch G, Ibáñez B, et al. Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC review topic of the week. J Am Coll Cardiol 2019;73(1):89-99.
[Crossref] [Google Scholar] [PubMed]
- Vinten‐johansen JA, Zhao ZQ, Nakamura M, Jordan JE, Ronson RS, Thourani VH, et al. Nitric oxide and the vascular endothelium in myocardial ischemia‐reperfusion injury. Ann N Y Acad Sci 1999;874(1):354-70.
[Crossref] [Google Scholar] [PubMed]
- Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol 2020;39(2):210-25.
[Crossref] [Google Scholar] [PubMed]
- Stamenkovic A, O’Hara KA, Nelson DC, Maddaford TG, Edel AL, Maddaford G, et al. Oxidized phosphatidylcholines trigger ferroptosis in cardiomyocytes during ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 2021;320(3):H1170-84.
[Crossref] [Google Scholar] [PubMed]
- Dong X, Qu S. Erigeron breviscapus (Vant.) hand-mazz.: A promising natural neuroprotective agent for alzheimer’s disease. Front Pharmacol 2022;13:1-13.
[Crossref] [Google Scholar] [PubMed]
- Liu G, Liang Y, Xu M, Sun M, Sun W, Zhou Y, et al. Protective mechanism of Erigeron breviscapus injection on blood-brain barrier injury induced by cerebral ischemia in rats. Sci Rep 2021;11(1):1-23.
[Crossref] [Google Scholar] [PubMed]
- Zhu J, Chen L, Qi Y, Feng J, Zhu L, Bai Y, et al. Protective effects of Erigeron breviscapus Hand.-Mazz.(EBHM) extract in retinal neurodegeneration models. Mol Vis 2018;24:315-25.
[Google Scholar] [PubMed]
- Liu H, Yang X, Ren T, Feng Y, Xu H. Effects of Erigeron breviscapus ethanol extract on neuronal oxidative injury induced by superoxide radical. Fitoterapia 2005;76(7-8):666-70.
[Crossref] [Google Scholar] [PubMed]
- Fang S, Dong L, Liu L, Guo J, Zhao L, Zhang J, et al. HERB: A high-throughput experiment-and reference-guided database of traditional Chinese medicine. Nucleic Acids Res 2021;49(D1):D1197-206.
[Crossref] [Google Scholar] [PubMed]
- Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C, et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res 2021;49(W1):W5-14.
[Crossref] [Google Scholar] [PubMed]
- Daina A, Michielin O, Zoete V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res 2019;47(W1):W357-64.
[Crossref] [Google Scholar] [PubMed]
- Otasek D, Morris JH, Bouças J, Pico AR, Demchak B. Cytoscape automation: Empowering workflow-based network analysis. Genome Biol 2019;20(1):1-5.
[Crossref] [Google Scholar] [PubMed]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47(D1):D607-13.
[Crossref] [Google Scholar] [PubMed]
- Tang Y, Li M, Wang J, Pan Y, Wu FX. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems 2015;127:67-72.
[Crossref] [Google Scholar] [PubMed]
- Yu G, Wang LG, Han Y, He QY. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012;16(5):284-7.
[Crossref] [Google Scholar] [PubMed]
- Liu Y, Grimm M, Dai WT, Hou MC, Xiao ZX, Cao Y. CB-Dock: A web server for cavity detection-guided protein-ligand blind docking. Acta Pharmacol Sin 2020;41(1):138-44.
[Crossref] [Google Scholar] [PubMed]
- Case DA, Cheatham III TE, Darden T, Gohlke H, Luo R, Merz Jr KM, et al. The Amber biomolecular simulation programs. J Comput Chem 2005;26(16):1668-88.
[Crossref] [Google Scholar] [PubMed]
- Lee TS, Allen BK, Giese TJ, Guo Z, Li P, Lin C, et al. Alchemical binding free energy calculations in AMBER20: Advances and best practices for drug discovery. J Chem Inf Model 2020;60(11):5595-623.
[Crossref] [Google Scholar] [PubMed]
- Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 2006;25(2):247-60.
[Crossref] [Google Scholar] [PubMed]
- Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high‐quality atomic charges. AM1‐BCC model: II. Parameterization and validation. J Comput Chem 2002;23(16):1623-41.
[Crossref] [Google Scholar] [PubMed]
- Tian C, Kasavajhala K, Belfon KA, Raguette L, Huang H, Migues AN, et al. ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J Chem Theory Comput 2019;16(1):528-52.
[Crossref] [Google Scholar] [PubMed]
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem 2004;25(9):1157-74.
[Crossref] [Google Scholar] [PubMed]
- Simmonett AC, Brooks BR. Analytical hessians for Ewald and particle mesh Ewald electrostatics. J Chem Phys 2021;154(10):104101.
[Crossref] [Google Scholar] [PubMed]
- Ryckaert JP, Ciccotti G, Berendsen HJ. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J Comput Phys 1977;23(3):327-41.
- Roe DR, Cheatham III TE. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput 2013;9(7):3084-95.
[Crossref] [Google Scholar] [PubMed]
- Dwivedi PS, Patil R, Khanal P, Gurav NS, Murade VD, Hase DP, et al. Exploring the therapeutic mechanisms of Cassia glauca in diabetes mellitus through network pharmacology, molecular docking and molecular dynamics. RSC Adv 2021;11(62):39362-75.
[Crossref] [Google Scholar] [PubMed]
- Dong Y, Chen H, Gao J, Liu Y, Li J, Wang J. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J Mol Cell Cardiol 2019;136:27-41.
[Crossref] [Google Scholar] [PubMed]
- Tibaut M, Mekis D, Petrovic D. Pathophysiology of myocardial infarction and acute management strategies. Cardiovasc Hematol Agents Med Chem 2016;14(3):150-9.
[Crossref] [Google Scholar] [PubMed]
- Wang Z, Yu J, Wu J, Qi F, Wang H, Wang Z, et al. Scutellarin protects cardiomyocyte ischemia-reperfusion injury by reducing apoptosis and oxidative stress. Life Sci 2016;157:200-7.
[Crossref] [Google Scholar] [PubMed]
- Xu LJ, Chen RC, Ma XY, Zhu Y, Sun GB, Sun XB. Scutellarin protects against myocardial ischemia-reperfusion injury by suppressing NLRP3 inflammasome activation. Phytomedicine 2020;68:1-11.
[Crossref] [Google Scholar] [PubMed]
- Shi M, Liu Y, Feng L, Cui Y, Chen Y, Wang P, et al. Protective effects of scutellarin on human cardiac microvascular endothelial cells against hypoxia-reoxygenation injury and its possible target-related proteins. Evid Based Complement Alternat Med 2015;2015:1-13.
[Crossref] [Google Scholar] [PubMed]
- Wang P, Huang H, Chen B, Su Y, Shi P, Yao H. Systems pharmacology dissection of mechanisms of dengzhan xixin injection against cardiovascular diseases. Chem Pharm Bull 2020;68(9):837-47.
[Crossref] [Google Scholar] [PubMed]
- Zhou SF, Yuan J, Liao MY, Xia N, Tang TT, Li JJ, et al. IL-17A promotes ventricular remodeling after myocardial infarction. J Mol Med 2014;92(10):1105-16.
[Crossref] [Google Scholar] [PubMed]
- Zhang A, Mao X, Li L, Tong Y, Huang Y, Lan Y, et al. Necrostatin‐1 inhibits Hmgb1‐IL‐23/IL‐17 pathway and attenuates cardiac ischemia reperfusion injury. Transpl Int 2014;27(10):1077-85.
[Crossref] [Google Scholar] [PubMed]
- DiDonato JA, Mercurio F, Karin M. NF‐κB and the link between inflammation and cancer. Immunol Rev 2012;246(1):379-400.
[Crossref] [Google Scholar] [PubMed]
- Shih RH, Wang CY, Yang CM. NF-kappaB signaling pathways in neurological inflammation: A mini review. Front Mol Neurosci 2015;8:1-8.
[Crossref] [Google Scholar] [PubMed]
- Latanich CA, Toledo-Pereyra LH. Searching for NF-κB-based treatments of ischemia reperfusion injury. J Invest Surg 2009;22(4):301-15.
[Crossref] [Google Scholar] [PubMed]
- Nichols TC. NF-kappaB and reperfusion injury. Drug News Perspect 2004;17(2):99-104.
[Crossref] [Google Scholar] [PubMed]
- Gao XM, Su Y, Moore S, Han LP, Kiriazis H, Lu Q, et al. Relaxin mitigates microvascular damage and inflammation following cardiac ischemia-reperfusion. Basic Res Cardiol 2019;114(4):1-5.
[Crossref] [Google Scholar] [PubMed]
- Raleigh JM, Toldo S, Das A, Abbate A, Salloum FN. Relaxin’the heart: A novel therapeutic modality. J Cardiovasc Pharmacol Ther 2016;21(4):353-62.
[Crossref] [Google Scholar] [PubMed]
- Thokala S, Inapurapu S, Bodiga VL, Vemuri PK, Bodiga S. Loss of ErbB2-PI3K/Akt signaling prevents zinc pyrithione-induced cardioprotection during ischemia/reperfusion. Biomed Pharmacother 2017;88:309-24.
[Crossref] [Google Scholar] [PubMed]
- Yuan X, Juan Z, Zhang R, Sun X, Yan R, Yue F, et al. Clemastine fumarate protects against myocardial ischemia reperfusion injury by activating the TLR4/PI3K/Akt signaling pathway. Front Pharmacol 2020;11:1-13.
[Crossref] [Google Scholar] [PubMed]
- Komar HM, Serpa G, Kerscher C, Schwoegl E, Mace TA, Jin M, et al. Inhibition of Jak/STAT signaling reduces the activation of pancreatic stellate cells in vitro and limits caerulein-induced chronic pancreatitis in vivo. Sci Rep 2017;7(1):1-10.
[Crossref] [Google Scholar] [PubMed]
- Bolli R, Dawn B, Xuan YT. Role of the JAK-STAT pathway in protection against myocardial ischemia/reperfusion injury. Trends Cardiovasc Med 2003;13(2):72-9.
[Crossref] [Google Scholar] [PubMed]
- Zhang Q, Wang L, Wang S, Cheng H, Xu L, Pei G, et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct Target Ther 2022;7(1):1-38.
[Crossref] [Google Scholar] [PubMed]
- Xuan YT, Guo Y, Han H, Zhu Y, Bolli R. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc Natl Acad Sci USA 2001;98(16):9050-5.
[Crossref] [Google Scholar] [PubMed]





