- *Corresponding Author:
- M. Sarfraz
Faculty of Pharmacy, ILM College of Pharmaceutical Sciences, Sargodha, Sargodha 40100, Pakistan
E-mail: chiefpharm@gmail.com
| Date of Received | 04 July 2021 |
| Date of Revision | 06 June 2023 |
| Date of Acceptance | 09 January 2024 |
| Indian J Pharm Sci 2024;86(1):32-44 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
Roflumilast, a selective phosphodiesterase 4 inhibitor, was approved by the European Medicines Agency Committee for Medicinal Products for Human Use in 2010 for the treatment of severe chronic obstructive pulmonary disease. However, low solubility of Roflumilast is a question mark to design a suitable dosage form to get therapeutic objectives. The aim of this work was to enhance solubility of the drug to develop fast release oral tablet. Drug-polymer binary system was developed at 1:1 w/w ratio followed by development of immediate release tablet by direct compression method. Drug-polymer compatibility studies were assessed by Fourier-transform infrared spectroscopy, X-ray Diffraction, thermogravimetric analysis, and scanning electron microscopic studies. Pre and Post-compression tests and in vitro dissolutions studies in acidic (pH 1.2) and PBS pH 6.8 were performed to evaluate in vitro performance of the developed formulations. In vitro kinetic models were applied and similarity and difference factors were computed in comparison with reference product (marketed brand). Drug content was found between 95.37 to 99.57 in the developed formulations. Drug-polymer compatibility results demonstrated good compatibility among drug, polymers and other excipients. The micromeretic parameters and post compression studies results were in acceptable limit for all the developed formulations. Fast drug release behavior was observed in acidic medium. The results suggested that similarity factor (f2) values of the developed formulations were within acceptable limit (50-100). The developed binary systems may apply to overcome solubility issues of biopharmaceutical classification system class II drugs. The developed formulations are easy to scale-up.
Keywords
Roflumilast, binary system, solid dispersion, chronic obstructive pulmonary disease, infrared spectroscopy tablet
Asthma and Chronic Obstructive Pulmonary Disease (COPD) are respiratory disorders that overlap on their clinical manifestation such as chest tightness, shortness of breath, cough and wheezing. However, bronchial asthma is an allergic disorder that is originated in childhood, physiologically characterized by reversible airflow obstruction, associated with eosinophil, and best responds to anti-inflammatory medication. In contrast, COPD is observed in mid or late life due to smoke, associated with neutrophils, responds to bronchodilators and avoid risk factors (chemicals, air pollutants, smog etc.), characterized by irreversible airflow limit leads to progress decline in lung function and ultimately early death[1]. However, spirometry results, after administration of bronchodilator, differentiate the both conditions (asthma and COPD), COPD is generally less respond to bronchodilator medication[2]. Inhaled glucocorticoids in combination with long acting bronchodilators was also failed to achieve therapeutic targets in COPD patients[3]. Moreover, air pollutants (O3, SO2, NO2, CO, PM2.5 and PM10) have been shown to exacerbate COPD associated illness[4]. Seasonal smog was also found an exacerbating element for COPD among the populations[5].
Roflumilast (3-cyclopropylmethoxy-4-difluoromethoxy-N-[3, 5-dichloropyrid-4-yl]-benzamide) is a selective effective PDE 4 inhibitor that brings new hope to cope with COPD[6-8]. Roflumilast and Roflumilast N-oxide are selective Phosphodiesterase 4 (PDE4), the PDE isoenzyme mainly found in inflammatory lung cells. At cellular level, PDE4 is responsible to convert cyclic Adenosine-3, 5-Monophosphate (cAMP) to the inactive 5’ nucleotide monophosphate. Thus, Roflumilast blocks the hydrolytic breakdown of (cAMP) to inactive 5-nucleotide monophosphate (AMP), which results in the intracellular accumulation of cAMP in target cell which is responsible to prevent the pathogenesis of COPD and inhibition of inflammatory cell activity[9]. PDE4 inhibitors are now considered by the clinicians a better option in severe COPD management[10].
60 %-70 % of the new drug molecules on market belong to Biopharmaceutical Classification System (BCS) class II drugs (low solubility, high permeability)[11]. Poor water solubility of these drugs restricts their oral bioavailability. Various solid dispersion techniques such as fusion method, melting method, kneading method, co-grinding (micronization) method, co-precipitation method, lyophilization method, congealing method, hot extrusion method, solvent evaporation method, supercritical fluid method etc. have been successfully employed to enhance dissolution of poorly water-soluble drugs in the bio-media of the body and hence improve bioavailability[12,13]. This solid dispersion technology throughputs fast production process of poorly water soluble drugs via improving drug loading, wettability, removal of crystallinity, and compressibility[13,14]. Amalgamation of drug moiety with polymeric solution improves stability, solubility and bioavailability of the drug[15].
80 % bioavailability of Roflumilast reported after oral administration and once daily oral dose is available in international market (DALIRESP®, DAXAS® etc.). However, to fulfill the demand of local market, we developed immediate release oral formulations of Roflumilast in collaboration with industry partner that can be easily scale up and can be manufactured at lowest cost. In this study, a binary system of Roflumilast and polymer (mannitol, Polyethylene Glycols (PEG) 6000, poloxamer 188 and gelucire 44/14) followed by immediate release tablet formulations was designed for the first time to address dissolution related issues of this drug. Core objective of this project was to see the impact of different polymers on drug release. Polymer selection was made on the basis of easy availability and low cost to reduce manufacturing cost for large scale production of the developed formulations by the partner industry in future after getting product registration approval from competent authority.
Materials and Methods
Materials:
Active pharmaceutical ingredient (Roflumilast) was donated by MCOLSON Research Laboratories Pvt. Ltd, Lahore, Pakistan, PEG 6000 (Merck, Germany), methanol (Emirgul Gida, Turkey), poloxamer 188 (Spectrum Chemical MFG Corp, USA), gelucire 44/14 (Gattefosse, USA), magnesium stearate (Fluka, Germany), talc (Zenith Mining and Construction, East Europe), primo gel (DFE pharma, Germany), lactose (BDH, England), starch (BDH, England). Distilled water was prepared in research laboratories of The University of Lahore.
Preparation of solid dispersion (binary system):
Roflumilast binary system was prepared at fixed ratio of drug and polymer (1:1, w/w) by kneading method. Briefly, the polymer (PEG 6000, mannitol, poloxamer and gelucire) was first melted at approximately 60° in China dish and drug was dispersed in molten mixture with constant stirring. The obtained solid dispersion was cooled under ice-water mixture for a period of 1 h. Each solid dispersion individually designated as SD1 (Roflumilast and PEG 6000), SD2 (Roflumilast and mannitol), SD3 (Roflumilast and poloxamer) and SD4 (Roflumilast and gelucire).
Solubility studies:
Solubility studies of pure drug and solid dispersions (SD1, SD2, SD3, and SD4) were carried out in methanol:water (1:1, v/v). Pure drug and solid dispersions were placed in eppendorf tubes containing 5 ml of solvent. The tubes were placed on shaking water bath at 37° for 1 h and centrifuged for 15 min at 6000 rpm. A Whattman filter paper was used to filter suspension and the filtrate was diluted appropriately and then analyzed spectrophotometrically (Shimadzu 1601, Japan) at 251 nm.
Fourier Transform Infrared Spectroscopy (FTIR) studies: FTIR (PerkinElmer, USA) spectroscopy was utilized to determine the spectra of drug, polymers and formulations to evaluate the interaction between drug and carrier in solid state by attenuated total reflectance (ATR) technique. The infrared spectra of drug, polymer and formulations were recorded from 400 and 4000 cm-1 with 10 times scanning.
X-Ray Diffraction (XRD) studies: XRD studies were performed to determine the powder characteristics of drug and solid dispersion formulations. Powder XRD patterns were recorded by (Meniflex 600 Rigaku, Japan) using Nifiltered Cuk α radiation with a voltage of 30 kV and a current of 25 mA. The scanning range was 4°-40° diffraction angle (2θ) at a scanning rate of 1°/min.
Thermogravimetric Analysis (TGA): TGA instrument ((Pyris Diamond Series TG/DTA, Perkin Elmer, USA) was employed to obtained TGA curves of Roflumilast, polymers and formulations. Approximately 5 mg of each sample on alumina crucible at a heating rate of 10°/min in the range of 40°-600° with flow rate 20 ml/min under nitrogen atmosphere.
Preparation of immediate release tablet:
Tablets were prepared by direct compression method using single punch tablet machine (Emmy, Pakistan). Briefly, pre-formulated binary systems (SD1/SD2/SD3/SD4) and excipients, such as lactose (83 % w/w), starch (quantity sufficient for 75 mg tablet weight), and primogel (5 % w/w), were homogenized for 10-20 min and placed in oven at 60° for 2 h. This homogenous mixture was passed through a sieve 180-mesh and uniform granules were obtained. After granulation, magnesium stearate and talcum powder (0.5 % w/w) were added to improve flow ability of the materials. The average weight of the tablets was 75 mg out of which 500 µg of pure Roflumilast and the rest was inactive materials. Each formulation designated individually with respect to their binary system such as F1 (formulation containing SD1), F2 (formulation containing SD2), F3 (formulation containing SD3), and F4 (formulation containing SD4).
Pre-compression tests:
The formulated granules were qualified for compression if met criteria of bulk density, Hausner’s ratio, tapped density, angle of repose and Carr’s index.
Angle of repose (0)=tan-1h/r (1)
Bulk density=M/Vb (2)
Tapped density=M/Vt (3)
Compressibility index=dt-db/dt×100 (4)
Hausner’s ratio=dt/db (5)
Where h: height of powder, r: Radius of powder, M: Mass of powder, Vb: Bulk volume of powder, Db: Bulk density of powder Dt : Tapped density of powder and Vt: Tapped volume of powder
Post-compression tests:
The post compression test of prepared tablets were evaluated for weight variation, hardness, thickness, friability, content uniformity and disintegration time according to method described in British Pharmacopeias. 20 tablets were selected randomly from each batch and weighed separately. For determination of weight variation, tablet individual weights were then compared with average weight. Tensile strength (Kg/cm²) factor is a compaction of tablet. Monsanto hardness tester and Vernier caliper were used to check the hardness and thickness of tablets.
Friability: 10 tablets from each lot were randomly selected and weighed. Ability of the tablets to withstand mechanical strength during manufacturing was measured by Roche friabilator (Curio, Pakistan) at 25 rpm (100 revolutions) for 4 min. Soft muslin cloth was used to dust off the tablet and reweighed. % Friability was measured by the formula:
Friability (%)=Tb-Ta/Tb×100 (6)
Where Tb and Ta are the weight of tablets before and after the test, respectively.
Content uniformity: Ten Roflumilast tablets were randomly selected, weighed and tablet powder equivalent to average weight of one tablet was transferred into 100 ml volumetric flask containing phosphate buffer (pH 6.8) and shake for 10 min. A supernatant solution was filtered through 0.45 µm filter and spectrophotometrically analyzed at 251 nm based on full UV scan at range of 400-200 nm (United States Pharmacopeia, 2011).
In vitro disintegration time: The disintegration test was carried out using the Disintegration apparatus (Pharmao Industries CO. LTD, China). Six tablets were selected randomly from each formulation for determination of disintegration time which is the time taken for the disintegration of tablets to the extent that no detectable mass remains in apparatus (United States Pharmacopeia, 2011).
Scanning Electron Microscopy (SEM): Surface morphology of tablets was determined by SEM (JSM-6380A, Joel, Japan). The formulation samples were mounted on the brass stub and coated with gold by auto coater (JFC-1500, Joel, Japan). Gold coated particles were placed in SEM chamber and scanning was performed at magnification range from 100-5000 at 10 kV voltage.
In vitro drug performance (dissolution study): Dissolution studies were performed in 0.1 N HCl (pH 1.2) and phosphate buffer (pH 6.8) by using USP type II apparatus (paddle method). The test was performed in 900 ml dissolution media with maintained temperature at 37.0°±.5° and paddle rotated at 75 rpm. Samples of 5 ml were taken at predetermined intervals through GD/XTM syringe filter and replaced with equal amount of fresh medium. Dissolution fluid was used to dilute the collected samples then all samples were assayed with UV spectrophotometer at preset conditions.
Zero order, first order and Higuchi models were employed to study release kinetics of the developed formulations by the following equations, respectively.
Qt=Q0+k0t (7)
logQ=logQ0=Kt/2.303 (8)
Qt=A√D(2Q0-Qs) Qst (9)
Where Qt is rate of drug release amount in time “t” and Q0 is initial drug amount in solution and K is zero order and first order rate constant. D is diffusivity of the drug molecules and Qs is solubility of drug in the matrix medium.
Model-independent approach was also used to analyze dissolution profiles by using dissimilarity (f1) and similarity factor (f2)[16].
f1= {∑ni=1 (Rj-Tj)/∑ni=1 Rj}×100 (10)
f2=50 log {1+(1/n) ∑ni=1 (Rj-Tj)²}-0.5×100 (11)
Where n is sample number, Rj and Tj are the percentages of dissolved reference and test products at time j respectively.
Statistics analysis:
GraphPad Prism 5 was used for statistics. Analysis of Variance (ANOVA) test was applied for group comparison.
Results and Discussion
Binary system of Roflumilast was prepared to improve solubility of the drug before preparing oral tablets. Methanolic water (50:50, v/v) was used to perform the solubility studies of solid dispersion. Solubility of pure Roflumilast was found 0.185±0.005 μg/ml. In comparison with pure Roflumilast, 1.87, 1.88, 1.91 and 1.87 fold increase in solubility was observed for the developed solid dispersions SD1, SD2, SD3 and SD4 respectively, as shown in fig. 1. Solubility of drug in Dispersion 1 (SD1) was increased, probably because of extensive hydrophobic interaction between PEG 6000 and drug. In dry form, particles of drug adhere with polymeric particles because of fusion method (shown in SEM). Aqueous channels/pore formation in solid dispersion with mannitol (SD2) led to increase in solubility of drug[17]. The developed binary system containing poloxamer 188 (SD3) exhibited higher solubility as compared with other formulations (1.91 folds) (fig. 1). Increase solubility of SD3 might be due to erosion and solubilization effect of polymer[18]. In SD4, Gelucire 44/14 destroyed the crystallinity of drug and presented suitable modification for enhancement of solubility[19].

Fig. 1: Solubility profile of Solid Dispersions (SD) of Roflumilast; SD1 (Roflumilast and PEG6000); SD2 (Roflumilast and Mannitol); SD3 (Roflumilast and Poloxamer 188); SD4 (Roflumilast and Gelucire) 44/14.
When these solid dispersion particles come in contact with solvent or water, the polymeric particles hydrate rapidly which contributed the increased wetting of drug particles. Enhanced solubility of solid dispersion of Roflumilast related to wetting effect and surface activity that leads towards increase in solubility effect, pore formation, surface area, reduction in agglomeration and transformation of crystalline to amorphous form[20].
FTIR spectra showed possible interaction between drug and polymer. Peaks for free Roflumilast observed at wave number: N-H stretching at 3255 cm-1, C=O stretching of amide moiety vibration at 1625 cm-1, C=C aromatic vibration at 1598 cm-1 and 1596 cm-1, -CH2- deformation vibration at 1484 cm-1, C-O-C symmetrical stretching vibration in cyclopropyl moiety at 1199 cm-1, -CF2- asymmetric vibration at 1145 cm-1, C-H aromatic vibration at 869 cm-1, as shown in fig. 2a. Peaks of PEG 6000 were observed at wave number: -CF2- stretching at 11.01 cm-1 and -C-H- bond at 842 cm-1. Peaks of mannitol were observed at wave number N-H at 3289 cm-1 and C-O-C at 1284 cm-1, as shown in fig. 2b and fig. 2c. FTIR spectrum of SD1 and SD2 exhibited peaks at wave number N-H stretching at 3288 cm-1, CH2 aliphatic symmetrical and asymmetrical stretching vibration at 2892 cm-1, C=O stretching of amide moiety vibration at 1652 cm-1, CH2 deformation vibration at 1484 cm-1, C-O-C symmetrical stretching vibration in cyclopropyl moiety at 1280 cm-1, and -CF2- asymmetric stretching at 1110 cm-1, as shown in fig. 2d and fig. 2e.

Fig. 2: FTIR spectra of developed binary system (solid dispersions, SD); (a): Roflumilast; (b): PEG 6000; (c): Mannitol; (d): Roflumilast and PEG6000, SD1 and (e): Roflumilast and Mannitol, SD.
Fig. 3a represents peak of free Roflumilast. Peaks of Poloxamer 188 was observed at wave number -CH2- at 2894 cm-1 and -CF2- at 1105 cm-1, as shown in fig. 3b. Peaks of Gelucire 44/14 was observed at wave number N-H; 3268 cm-1 and -C=O-; 1650 cm-1, as shown in fig. 3c. Drug and polymer peaks also could be seen in the relevant spectrum of binary systems (SD3 and SD4), as shown in fig. 3d and fig. 3e, respectively.

Fig. 3: FTIR spectra of developed binary system (solid dispersions, SD); (a): Roflumilast; (b): Poloxamer 188; (c): Gelucire 44/14; (d): Roflumilast and Poloxamer, SD3 and (e): Roflumilast and Gelucire, SD4.
FTIR spectra of solid dispersions showed distinctive peaks with no apparent shifting which shows the absence of any chemical interactions[6]. Crystalline nature of drug, polymers and formulations were determined by XRD studies. XRD pattern of Roflumilast (fig. 4a) exhibited sharp peaks at a diffraction angle (20) of 6.00°, 17.05°, 23.15° and 24.15° and their respective counts were 308, 120, 175 and 493 respectively. PEG 6000 (fig. 4b) at diffraction angle (2θ) showed high intensity of 19.15° and 24.00° and their respective counts were 3125 and 340. Mannitol (fig. 4c) showed intense peaks at diffraction angle (2θ) of 14.60°, 19.10°, 22.95°, 29.40° and 24.65° and their respective counts were 2765, 390, 407, 1842 and 417 respectively. SD1 (fig. 4d) showed two peaks at diffraction angle (2θ) of 5.65° and 22.45° and their respective counts were 1949 and 1454. SD2 (fig. 4e) showed three peaks at diffraction angle (2θ) of 6.00°, 19.00° and 23.35° and their respective counts were 373, 250 and 2164, respectively.

Fig. 4: X-ray Diffraction analysis of developed binary systems (solid dispersion, SDs); (a): Roflumilast; (b): PEG 6000; (c): Mannitol; (d): Roflumilast and PEG6000, SD1 and (e): Roflumilast and Mannitol, SD2.
Fig. 5a represents free Roflumilast. Poloxamer 188 at diffraction angle (2θ) showed high intensity peaks of 19.15° and 23.30° and their respective counts were 2387 and 2698 respectively (fig. 5b). The gelucire 44/14 was confirmed by two crystalline peaks at 19.1° and 23.1° (fig. 5c). SD3 (fig. 5d) showed two peaks at diffraction angle (2θ) of 5.55° and 23.05° and their counts were 5891 and 555, respectively. SD4 (fig. 5e) confirmed two peaks at diffraction angle (2θ) of 5.9° and 23.45° with their respective counts were 5390 and 540, respectively.

Fig. 5: X-ray diffraction analysis of developed binary systems (solid dispersion, SDs); (a): Roflumilast; (b): Poloxamer 188; (c): Gelucire 44/14; (d): Roflumilast and Poloxamer, SD3 and (e): Roflumilast and Gelucire, SD4.
The intensity of Roflumilast peaks was sharpen as shown in diffractogram of pure drug (fig. 4a and fig. 5a) but when mixed with polymers, loss of sharpness was observed that might be due to decrease in crystallinity, which revealed amorphous nature of solid dispersions[21].
Two mass loss patterns were seen in physical mixture of TG analysis. The first loss of mass was 10 %-20 % between 250°-270°, might be due to evaporation of absorbed water. The second mass loss was observed 65 %-70 % at 370°-490°, due to slow break down of Roflumilast (fig. 6 and fig. 7).

Fig. 6: Thermograms of developed binary systems; SD1 (Roflumilast and PEG6000) and SD2 (Roflumilast and Mannitol).
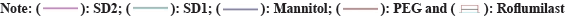 .
.

Fig. 7: Thermograms of developed binary systems: SD3 (Roflumilast and Poloxamer), SD4 (Roflumilast and Gelucire).
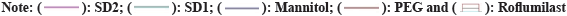 .
.
Maximum loss of mass of each polymer was found within temperature range of 200°-600° as shown in TG data. Heat given out by melting of pure drug, in most of the cases was protected with least changes of boarding or shifting towards the lower temperature. The peak shape and enthalpy value in drug polymer mixture appears to be affected by the quantity of material used. Hence, pure drug and polymers mixtures may be cause of minor changes in heat given out by melting of drug as the process of mixing alters the purity of each component of mixture[6].
Physical attributes of formulated granules were tested. Angle of repose less than or equal to 40° indicates free flowing properties of powder. The results of angle of repose showed good to acceptable flow properties of granules[22]. Angle of repose was measured, which was 26°±2.64°, 27°±1.52°, 28°±2.00° and 29°±2.64° for F1, F2, F3 and F4, respectively (Table 1). The overall bulk density was also in acceptable range 0.47±0.02-0.58. The bulk density values less than 1.2 g/cm3 indicated good flow properties and values greater than 1.5 g/cm3 specify poor flow rate. The tapped density values were in the range of 0.53±0.036 to 0.65±0.025 g/cm3 (Table 2). Bulk and tapped densities of developed formulations provides significant information about packing properties. The compressibility index was in range between 10.76±2.436 to 15.87±2.579 that supported of good flow property[23]. Similarly, the values of Hausner’s ratio were in the range of 1.12±1.147 to 1.18±1.152 which was less than 1.25 which indicate fair flow properties. The flow properties of formulated granules were found to be greater than formulation ingredients due to their spherical and smooth surface. It was confirmed that polymeric material contributes to improve the flow properties and used for slope determination. Results revealed that all formulations expressed excellent flow characteristics that were within prescribed pharmacopeial limits (Table 1).
| Formulations | Bulk density (g/cm³) | Tapped density (g/cm³) | Angle of repose (θ) | Carr’s index (Ci) | Hausner’s ratio |
|---|---|---|---|---|---|
| F1 | 0.47±0.02 | 0.53±0.036 | 26°±2.64° | 11.32±1.863 | 1.12±1.147 |
| F2 | 0.50±0.04 | 0.59±0.035 | 27°±1.52° | 15.25±2.905 | 1.18±1.137 |
| F3 | 0.58±0.07 | 0.65±0.025 | 28°±2.00° | 10.76±2.436 | 1.12±1.117 |
| F4 | 0.53±0.09 | 0.63±0.035 | 29°±2.64° | 15.87±2.579 | 1.18±1.152 |
Table 1: Pre-Compression attributes of developed formulations.
| Formulations | Weight variation (mg) | Thickness (mm) | Hardness (kg/cm²) | Friability | Disintegration time (s) | Content uniformity (%) |
|---|---|---|---|---|---|---|
| F1 | 74.80±0.26 | 2.33±0.05 | 2.4±0.1 | 0.54±0.03 | 63±0.03 | 97.34±1.3 |
| F2 | 74.56±0.45 | 2.54±0.15 | 2.6±0.2 | 0.56±0.03 | 73±0.02 | 98.26±1.4 |
| F3 | 74.80±0.26 | 2.51±0.20 | 2.8±0.3 | 0.92±0.04 | 90±0.04 | 95.37±1.2 |
| F4 | 74.83±0.32 | 2.46±0.10 | 2.9±0.1 | 0.87±0.04 | 87±0.03 | 99.57±1.3 |
Table 2: Post-Compression attributes of developed formulations.
Developed formulations were characterized for weight variation, hardness, thickness, friability, and content uniformity (Table 2). The average weight variation of 10 tablets of each formulation was less than 7.5 %. The hardness of tablets from all formulation were found to be in range from 2.4±0.1 to 2.9±0.1 and the thickness of tables from all batches ranged between 2.33±0.05 to 2.54±0.15. Tablets of all batches showed uniform thickness.
Percentage friability of all developed formulations was found in range of 0.54±0.03 to 0.92±0.04 which indicates that the friability is within the range as the weight loss was below 1 % (Table 2). Friability results suggested all prepared tablets have good mechanical resistance and may not break throughout handling on machines and during shipping.
Percentage of active drug was in range of 95.37 to 99.57 in the developed formulations. The variation was found to be less than 1.4 %, which indicated good drug content uniformity in prepared tablets (Table 2)[24].
The rapid disintegration assists swallowing and additionally plays a function in drug absorption in stomach, therefore promoting bioavailability. Disintegration time of prepared tablets vary from 63-90 s because the concentration of disintegrant within formulations decreases the disintegration time (Table 2)[25].
Morphology of developed formulations can be observed by SEM images (fig. 8). Polymer play vital role in shape and geometry of their blended mixtures. Compression force transformed shape and geometry of the mixture lead to smooth surface of resultant compact mass under the influence of surface tension and thermal expansion. SEM images revealed crystal formation in the developed formulations. Layered crystals were observed in F1 formulation showing flake arrangement in shape and geometry. F2 formulation found with smoother surface. F3 formulation was in rocky appearance with rough surface. F4 formulation showed fused and compact mass with groove-patterned surface. This diversity in surface morphology is definitely due to physicochemical properties of different polymers that were used in these formulations. However, surface coating can give smoothness and elegant appearance to developed tablet formulations. Studies demonstrated impact of molecular weight and physicochemical properties of polymer influence surface morphology[26-29].

Fig. 8: Morphological studies of developed tablets by scanning electron microscopy; (a): Formulation 1; (b): Formulation 2; (c): Formulation 3; (d): Formulation 4; upper row: 100X magnification and lower row: 1500X magnification.
Immediate release formulations having ideal dissolution conditions generally release 100 % of active pharmaceutical product within 60-90 min. Comparative in vitro drug release study of developed formulations and reference product Rofin® (MCOLSON Research Lab, Pakistan) was conducted in acidic medium at pH 1.2 (0.1 N HCl) and phosphate buffer pH 6.8.
In acidic medium, 70 %-80 % drug release was observed after 45 min from all the developed formulations and reference product (Rofin®). After 90 min, the drug release was 80 %-90 %. More than 90 % drug release was achieved after 120 min. Drug release profile of developed formulations in acidic medium is shown in fig. 9a. In phosphate buffer (pH 6.8), F2, F3 and F4 formulations showed 40 %-45 % release of drug within 45 min while F1 and Rofin® showed more than 50 % drug release. However, after 90 min, the release of drug from F1, F2 and Rofin® was more than 70 % but the drug release from F3 and F4 formulations was restricted to less than 70 %. After 120 min, formulations F2, F3 and Rofin® showed drug release up to 90 %. Formulations F1 and F4 showed highest drug release at pH 6.8 that was more than 90 % after 120 min. Drug release profile of developed formulations buffer at pH 6.8 is shown in fig. 9b.

Fig. 9: In vitro performance (dissolution study) of developed formulations; (a): Acidic medium pH 1.2 (0.1 N HCl) and (b):
Phosphate buffer pH 6.8.
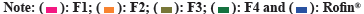 .
.
Like marketed reference product, drug release from all the developed formulations was very fast in acidic medium i.e. 40 %-50 % drug was released within 5 min. Although only 10 % drug release was observed in buffer medium at pH 6.8.
In vitro dissolution studies of Roflumilast immediate release tablet suggested that increased rate of dissolution was due to solubilization effect of polymeric carrier, dispersibility, improvement in wetting by carrier, de-aggregation of drug crystals, drug dissolution with carrier and conversion of drug crystalline nature into amorphous form. It is known that drug in amorphous form exhibits fast dissolution rate. Significant difference in drug release among developed formulations is shown in Table 3.
| Group | 0.1 N HCl | pH 6.8 |
|---|---|---|
| F1 vs. F2 | No | No |
| F1 vs. F3 | No | No |
| F1 vs. F4 | No | No |
| F1 vs. Rofin® | No | No |
| F2 vs. F3 | No | No |
| F2 vs. F4 | Yes** | No |
| F2 vs. Rofin® | No | No |
| F3 vs. F4 | No | No |
| F3 vs. Rofin® | No | No |
| F4 vs. Rofin® | Yes** | Yes** |
Note: (*): Significant p<0.05
Table 3: Statistical difference in drug release profile of developed formulations.
Firstly, model dependent methods were used to analyze the data from dissolution test. Three kinetic models i.e. first order, zero order and Higuchi were applied to evaluate drug release from reference product Rofin® and formulated tablets. All formulations (F1, F2, F3, F4 and Rofin®) followed first order kinetics and showing highest R2 values i.e. 0.4434, 0.4187, 0.7196, 0.5576 and 0.8361 respectively in 0.1 N HCl (pH 1.2) which indicate that these formulations were dependent on initial concentration. At phosphate buffer (pH 6.8), three formulations (F1, F2 and F3) were followed first order kinetics and depicted highest R2 values i.e. 0.9936, 0.9843 and 0.9722 respectively. But reference product Rofin® and F4 formulation were followed zero order kinetic and showed highest R2 values i.e.0.9825 and 0.9752 respectively, which means it was independent of initial concentration[21,26]. In vitro release kinetics is given in Table 4.
| In vitro kinetic model | F1 | F2 | F3 | F4 | Rofin® |
|---|---|---|---|---|---|
| Drug release kinetics at 0.1 N HCl (pH 1.2) | |||||
| Zero order (R²) | -1.2154 | -1.5358 | -1.0286 | -0.7384 | -0.8848 |
| First order (R²) | 0.4434 | 0.4187 | 0.7196 | 0.5576 | 0.8361 |
| Higuchi model (R²) | 0.2774 | 0.0844 | 0.3744 | 0.4989 | 0.4555 |
| Drug release kinetics at phosphate buffer (pH 6.8) | |||||
| Zero order (R²) | 0.8696 | 0.9314 | 0.9403 | 0.9752 | 0.9825 |
| First order (R²) | 0.9936 | 0.9843 | 0.9722 | 0.9727 | 0.9717 |
| Higuchi model (R²) | 0.9795 | 0.9558 | 0.9332 | 0.8939 | 0.9675 |
Table 4: Drug release kinetics at 0.1 N HCL (PH 1.2) and phosphate buffer (pH 6.8).
Dissolution profiles can be compared on the basis of similarities (f2) and difference (f1) factor (model independent approach). The values of similarity and difference factor obtained from the comparison of dissolution profiles of Rofin® vs. F1 were 51.87 and 14.46, respectively, while for Rofin® vs. F2 were 54.53 and 13.44 respectively. The values of similarity and difference factor between Rofin® vs. F3 were 61.59 and 10.93, respectively and while for Rofin® vs. F4 were 48.16 and 32.72 respectively. The results suggested that similarities (f2) values of reference product (Rofin®) and test formulations (F1, F2, F4 and F3) were within acceptable limit (50-100) as shown in Table 5. However, significant difference in release behavior among F2 and F4 or F4 and Rofin® was might be due to use of different polymer in different formulations and each polymer has its own intrinsic behavior of drug release at different pH.
| Formulations | Similarity factor (f1) | Difference factor (f2) |
|---|---|---|
| Rofin® vs. F1 | 51.47 | 14.46 |
| Rofin® vs. F2 | 54.53 | 13.44 |
| Rofin® vs. F3 | 61.59 | 10.93 |
| Rofin® vs. F4 | 48.16 | 32.72 |
Table 5: Similarity (f1) and dissimilarity (f2) values of different dissolution profile.
In brief, PEG 6000 improved physical texture of the blended mixture, reduced disintegration time and increased solubility rate via increasing porosity and water influx. It also decreased degree of crystallinity of the corresponding SD1. Mannitol gave unique texture to its corresponding drug-polymer mixture (SD2) such as improved porosity, larger surface area, surface smoothness and increased molecular interlocking, ultimately improved tablet ability. Poloxamer 188 reduced surface tension and improved packing efficiency resulted in fast-release smooth-surface tablets (F3). Gelucire 44/14 improved solubilization of Roflumilast (SD4).
Stabilization and solubilization are the initial challenges that are faced by the formulation scientist during product development process for new drug moiety or new dosage form. Polymeric carrier system improves colloidal dispersion of poorly-water soluble drugs by reducing surface tension. In current study, we employed various polymers to enhance solubility of Roflumilast (BCS class II drug) that led to improve in vitro performance of developed immediate release tablets. The developed formulations of Roflumilast were compared with the available international brand Rofin® to observe any difference in drug release, if any. However, no significance difference was observed. Hence, we can claim that the used polymers are also suitable to design IR product of Roflumilast. Solubility of other drug candidates of BCS class II also can be improved if follow reported methodology per established criteria. Moreover, our laboratory findings provide basis to scale-up developed formulations for large scale production.
Conflict of interests:
The authors declared no conflict of interests.
References
- Kim SR, Rhee YK. Overlap between asthma and COPD: Where the two diseases converge. Allergy Asthma Immunol Res 2010;2(4):209-14.
[Crossref] [Google Scholar] [PubMed]
- Sim YS, Lee JH, Lee WY, Suh DI, Oh YM, Yoon JS, et al. Spirometry and bronchodilator test. Tuberc Respir Dis 2017;80(2):105-12.
[Crossref] [Google Scholar] [PubMed]
- Magnussen H, Disse B, Rodriguez-Roisin R, Kirsten A, Watz H, Tetzlaff K, et al. Withdrawal of inhaled glucocorticoids and exacerbations of COPD. New Engl J Med 2014;371(14):1285-94.
[Crossref] [Google Scholar] [PubMed]
- Li J, Sun S, Tang R, Qiu H, Huang Q, Mason TG, et al. Major air pollutants and risk of COPD exacerbations: A systematic review and meta-analysis. Int J Chron Obstruct Pulmon Dis 2016;11:3079-91.
[Crossref] [Google Scholar] [PubMed]
- Pothirat C, Tosukhowong A, Chaiwong W, Liwsrisakun C, Inchai J. Effects of seasonal smog on asthma and COPD exacerbations requiring emergency visits in Chiang Mai, Thailand. Asian Pac J Allergy Immunol 2016;34(4):284-9.
[Google Scholar] [PubMed]
- Ali F, Kumar R, Sahu PL, Singh GN. Physicochemical characterization and compatibility study of roflumilast with various pharmaceutical excipients. J Therm Anal Calorim 2017;130:1627-41.
- Boswell-Smith V, Spina D. PDE4 inhibitors as potential therapeutic agents in the treatment of COPD-focus on roflumilast. Int J Chron Obstruct Pulmon Dis 2007;2(2):121-9.
[Google Scholar] [PubMed]
- Brown WM. Treating COPD with PDE 4 inhibitors. Int J Chron Obstruct Pulmon Dis 2007;2(4):517-33.
- Tashkin DP. Roflumilast: The new orally active, selective phophodiesterase-4 inhibitor, for the treatment of COPD. Expert Opin Pharmacother 2014;15(1):85-96.
[Crossref] [Google Scholar] [PubMed]
- Rabe KF. Update on roflumilast, a phosphodiesterase 4 inhibitor for the treatment of chronic obstructive pulmonary disease. Br J Pharmacol 2011;163(1):53-67.
[Crossref] [Google Scholar] [PubMed]
- Chen A, Shi Y, Yan Z, Hao H, Zhang Y, Zhong J, Hou H. Dosage form developments of nanosuspension drug delivery system for oral administration route. Curr Pharm Des 2015;21(29):4355-65.
[Crossref] [Google Scholar] [PubMed]
- Zhang X, Xing H, Zhao Y, Ma Z. Pharmaceutical dispersion techniques for dissolution and bioavailability enhancement of poorly water-soluble drugs. Pharmaceutics 2018;10(3):74.
[Crossref] [Google Scholar] [PubMed]
- Tran P, Pyo YC, Kim DH, Lee SE, Kim JK, Park JS. Overview of the manufacturing methods of solid dispersion technology for improving the solubility of poorly water-soluble drugs and application to anticancer drugs. Pharmaceutics 2019;11(3):132.
[Crossref] [Google Scholar] [PubMed]
- Huang Y, Dai WG. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm Sin B 2014;4(1):18-25.
[Crossref] [Google Scholar] [PubMed]
- Marosi G. Editorial corner–a personal view: Polymer systems for solid pharmaceuticals. Express Polym Lett 2008;12(6):391.
- Emami J. In vitro-in vivo correlation: From theory to applications. J Pharm Pharm Sci 2006;9(2):169-89.
[Google Scholar] [PubMed]
- Wan S, Sun Y, Qi X, Tan F. Improved bioavailability of poorly water-soluble drug curcumin in cellulose acetate solid dispersion. AAPS PharmSciTech. 2012;13(1):159-66.
[Crossref] [Google Scholar] [PubMed]
- Lee KR, Kim EJ, Seo SW, Choi HK. Effect of poloxamer on the dissolution of felodipine and preparation of controlled release matrix tablets containing felodipine. Arch Pharm Res 2008;31:1023-8.
[Crossref] [Google Scholar] [PubMed]
- Alladi S, Shastri NR. Semi solid matrix formulations of meloxicam and tenoxicam: An in vitro and in vivo evaluation. Arch Pharm Res 2015;38:801-12.
[Crossref] [Google Scholar] [PubMed]
- Newa M, Bhandari KH, Oh DH, Kim YR, Sung JH, Kim JO, et al. Enhanced dissolution of ibuprofen using solid dispersion with poloxamer 407. Arch Pharm Res 2008;31(11):1497-507.
[Crossref] [Google Scholar] [PubMed]
- Farooq M, Shoaib MH, Yousuf RI, Qazi F, Hanif M. Development of extended release loxoprofen sodium multiparticulates using different hydrophobic polymers. Polym Bull 2019;76:2537-58.
- Elkhodairy KA, Hassan MA, Afifi SA. Formulation and optimization of orodispersible tablets of flutamide. Saudi Pharm J 2014;22(1):53-61.
[Crossref] [Google Scholar] [PubMed]
- Kumar PH, Bikshapathi D. Formulation and evaluation of fast dissolving tablets of Roflumilast solid dispersion. World J Pharm Sci 2015;4(8):1817-45.
- Leonardi D, Barrera MG, Lamas MC, Salomón CJ. Development of prednisone: Polyethylene glycol 6000 fast-release tablets from solid dispersions: Solid-state characterization, dissolution behavior, and formulation parameters. AAPS PharmSciTech 2007;8:221-8.
[Crossref] [Google Scholar] [PubMed]
- Zhao K, Yuan Y, Wang H, Li P, Bao Z, Li Y. Preparation and evaluation of valsartan by a novel semi-solid self-microemulsifying delivery system using Gelucire 44/14. Drug Dev Ind Pharm 2016;42(10):1545-52.
[Crossref] [Google Scholar] [PubMed]
- Wray P, Chan KA, Kimber J, Kazarian SG. Compaction of pharmaceutical tablets with different polymer matrices studied by FTIR imaging and X-ray microtomography. J Pharm Sci 2008;97(10):4269-77.
[Crossref] [Google Scholar] [PubMed]
- El-Bagory IM, Brakat N, El-Badry M, Ibrahim MA, El-Enazi F. Effect of polymer blend on diltiazem HCL matrix tablets prepared by direct compression. J Pharm Sci Technol 2010.
- Khadka P, Ro J, Kim H, Kim I, Kim JT, Kim H, et al. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J Pharm Sci 2014;9(6):304-16.
- Saeed MA, Ansari MT, Ch BA. Enhancement of solubility and dissolution profile of artesunate by employing solid dispersion approach: An in vitro evaluation. Pak J Pharm Sci 2019;32(1):33-61.
[Google Scholar] [PubMed]





