- *Corresponding Author:
- G. B. Preethi
Department of Pharmaceutics, Karnataka Lingayat Education College of Pharmacy, Bengaluru, Karnataka 560010
E-mail: preethigb_100@yahoo.com
| Date of Received | 22 May 2021 |
| Date of Revision | 01 October 2021 |
| Date of Acceptance | 06 June 2022 |
| Indian J Pharm Sci 2022;84(3):712-722 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
Etravirine is an antiviral belonging to biopharmaceutics classification system class IV due to low aqueous solubility and poor permeability. In this study, to augment the dissolution profile of etravirine, solid lipid nanoparticles have been formulated by utilizing lipid mixture of Compritol® 888 ATO and Gelucire® 50/13 as lipids and poloxamer 188 as surfactant by hot homogenization technique. Effect of variables, ratio of lipids, the concentration of surfactant and sonication time on responses like: Percentage yield, percentage encapsulation efficiency and percentage drug release were studied at 2 different levels and were statistically analyzed using 23 full factorial statistical design. The effect of factors on responses was well explained by a significant linear model. The optimized solid lipid nanoparticles preparation was evaluated for zeta potential, size of the particle and polydispersity index. The solid lipid nanoparticles displayed a zeta potential of 36.2 mV, the particle size of 178 nm and polydispersity index value of 0.075 indicating the solid lipid nanoparticles were nano sized and monodispersed. The absence of interaction of drug with excipients was confirmed with differential scanning calorimetry and X-ray powder diffractometer. In vitro drug profile of optimized solid lipid nanoparticles formulation was found to be 43.68 % in comparison with pure drug (19.99 %) and the release kinetics for optimum preparation was explained by first-order kinetics. Based on promising in vitro results it can be concluded that solid lipid nanoparticles of etravirine can be formulated based on validated factorial design, which may be a potential drug delivery system in the management of human immunodeficiency virus infection.
Keywords
Etravirine, solid lipid nanoparticles, 23 full factorial design, biopharmaceutics classification system class IV, antiretroviral drugs
Nanotechnology offers many advantages like targeted drug delivery and controlled/programmed release of therapeutic compounds for improved drug pharmacokinetics, biodistribution and pharmacodynamics activity. In addition, the strategy can be implemented to drugs belonging to Biopharmaceutics Classification System (BCS) class II and IV to increase their solubility and permeation through Gastrointestinal (GI) barrier. It can be used to solve the problems associated with poorly soluble drugs, thereby increasing the solubility and bioavailability[1,2]. In recent years lipid-based nanoparticles for drug delivery have gained attention due to their physiochemical diversity, biocompatibility and ability to enhance oral bioavailability of poorly water-soluble drugs[3,4]. Matrix type lipid-based Solid Lipid Nanoparticles (SLN) represent an alternative carrier system to conventional colloidal systems such as emulsions, liposomes and polymeric micro and nanoparticles. SLN are sub-micron colloidal carriers with particle size ranging from 50-1000 nm. SLN is biocompatible and biodegradable and has been used for controlled drug delivery and specific targeting. It also offers a greater surface area, prolonged drug release and instant uptake by the cells. It has the potential to overcome the solubility and bioavailability problem of poorly soluble drugs and enhances the bioavailability of entrapped drugs by enhancing the rate of dissolution and tissue distribution[5,6]. It can be administered by various routes and can incorporate hydrophilic/ hydrophobic drugs with different physicochemical and pharmacological properties[7,8].
Antiretroviral (ARV) drugs are clinically accepted for effective management of Acquired Immune Deficiency Syndrome (AIDS) by controlling of Human Immunodeficiency Virus (HIV). Etravirine (ETR) is a Non-Nucleoside Reverse Transcriptase Inhibitor (NNRTI) developed for the treatment of HIV-1 infection. It belongs to BCS class IV compound with low solubility and low permeability. With LogP>5, it is practically insoluble over a wide range of physiological pH and metabolized by cytochrome P450[9,10].
Novel drug delivery system has taken a new direction to reach the goals of ARV therapy in the management of HIV/AIDS[11]. SLN has been explored for antiviral drugs for targeting the lymphatic system[12-14] to enhance oral bioavailability by resolving solubility issues and to minimize drug toxicity and extensive first-pass metabolism. Therefore, these SLN holds great promise for reaching the goal of controlled and site-specific drug delivery.
Lipids in SLN preparation should be biocompatible and remain solid at body temperature. Compritol® 888 ATO (COM) has gained attention in the preparation of SLN for its ability to solubilize and entrap lipophilic drug due to its complex structure which gives more space for drug loading[15]. Gelucire® 50/13 (GEL) chemically named as stearoyl polyoxyl-32 glycerides, is a lipid that can enhance drug solubility and bioavailability due to its unique composition. It is blended with other lipids in SLN drug delivery system, for stabilizing lipid nano system and to increase drug loading[16-18].
Since not much work is reported on ETR-SLN preparation, in this work ETR-SLN was prepared using lipids COM and GEL, based on 23 factorial design and to evaluate the effect of variables on in vitro responses.
Materials and Methods
ETR was obtained as a generous gift sample from Apotex Research Private Limited (Bengaluru, India). GEL and COM are gift samples obtained from Gattefosse India Private Limited (Mumbai, India), poloxamer 188 from Sigma Aldrich, and the rest of the analytical grade chemicals and reagents were procured from Merck (Mumbai, India) and SD Fine Chem Limited (Mumbai, India).
Solubility study of ETR in lipids:
The specific amount of lipid was taken in a vial and melted at a temperature greater than its melting point on a hot plate. ETR was added in increments of 10 mg to the melted lipid and vortexed until the complete ETR was solubilized in the molten lipid. This is continued till the saturation level. The amount of drug solubilized in the lipid was determined analytically. Solid lipids such as COM, GEL, stearic acid and glyceryl monostearate were screened to determine the saturation solubility of ETR[13-16].
Formulation of SLN:
Hot homogenization technique was followed for preparation of SLN followed by probe ultra-sonication. The SLN formulation was prepared using GEL and COM as lipids and poloxamer 188 as surfactant. A weighed amount of solid lipids were taken and melted at 70° on a hot plate and ETR was dissolved in a molten lipid mixture. An aqueous phase was prepared separately in a beaker by dissolving poloxamer 188 in distilled water and heated up to 70°. Once the lipid mixture melts the hot surfactant aqueous solution was added to the molten lipid, it is homogenized at 15 000 rpm for 10 min in IKA T18 digital Ultra-Turrax. During this homogenization process, the temperature was maintained constant at 70°. The resultant oil/water (o/w) emulsion was ultrasonicated by probe sonicator-PUS-250 at 100 W for 5 and 10 min[12,19,20]. The obtained milky nano emulsion was cooled down to room temperature to form SLN suspension. The SLN suspension was centrifuged at 14 000 rpm and the supernatant solution was separated. After centrifugation, the dispersion of SLN is stored in an airtight container and dried in a vacuum desiccator.
ETR-SLN formulations by 23 full factorial design:
ETR-SLN formulations were prepared as per 23 factorial design. The independent variables selected were lipid ratio (A), surfactant concentration (B) and sonication time (C). Percentage (%) yield (Y1), % Entrapment Efficiency (% EE) (Y2), and % Drug Release (% DR) (Y3) were taken as the response parameters and are considered as dependent variables. Levels of independent variables are shown in Table 1 and the codes +1 and -1 indicates the high and low values of the independent variables. A total of eight formulations ET1 to ET8 with two levels of each factor were prepared according to a 23 full factorial experimental as shown in Table 2.
| Factor | Coded value | |
|---|---|---|
| -1 | +1 | |
| Lipid ratio (g) | 1:1 | 1:2 |
| Surfactant concentration | 1 % | 2 % |
| Sonication time | 5 min | 10 min |
Table 1: Independent variables and levels
| Formulation | ET1 | ET2 | ET3 | ET4 | ET5 | ET6 | ET7 | ET8 |
|---|---|---|---|---|---|---|---|---|
| Drug, ETR (mg) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| COM:GEL (g) | 1:1 (-1) | 1:1 (-1) | 1:2 (+1) | 1:2 (+1) | 1:1 (-1) | 1:2 (+1) | 1:1 (-1) | 1:2 (+1) |
| Poloxamer 188 (%) | 1 (-1) | 1 (-1) | 2 (+1) | 2 (+1) | 2 (+1) | 1 (-1) | 2 (+1) | 1 (-1) |
| Distilled water (ml) | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| Sonication time (min) | 5 (-1) | 10 (+1) | 10 (+1) | 5 (-1) | 10 (+1) | 5 (-1) | 5 (-1) | 10 (+1) |
Table 2: List of SLN formulations with coded values
Physicochemical characterization of SLN:
Percentage yield and % EE of ETR-SLN: The % yield (% Y) of the dried ETR-SLN was calculated by determining actual yield and theoretical yield[21]. Where actual yield is the amount recovered after preparation and theoretical yield is the yield calculated based on ingredients added in preparation. The % EE of ETRSLN was determined by the centrifugation method[13]. A fixed quantity of SLN dispersion was centrifuged at 20 000 rpm (Refrigerated micro centrifuge ELTEK RC 4815 F) for 25 min and the sample from supernatant solution was diluted using Dimethyl Sulfoxide (DMSO) and further quantified for free ETR using Ultraviolet- Visible (UV-VIS) spectrophotometer (Shimadzu UVVIS 1800 spectrophotometer) at 315 nm.
% EE=Weight of initial drug-Weight of free drug/ Weight of initial drug×100
In vitro drug release of ETR from ETR-SLN: In vitro drug release from ETR-SLN was performed by United States Pharmacopeia (USP) apparatus II (Paddle type) using 900 ml of 0.01 M Hydrochloric acid (HCl) with 1 % Sodium Lauryl Sulfate (SLS) at 37°±0.5° and 50 rpm. An aliquot of sample was withdrawn at regular time intervals of 30 min and after each sampling, the equivalent volume is replaced with fresh media and sample volume is filtered by 0.45 μm membrane filter[19]. The dissolution was performed for 8 h and the amount of drug released was analyzed using a UV-VIS spectrometer at 315 nm.
Statistical analysis and model validation: The responses obtained after in vitro evaluation were subjected to Design-Expert 12.0 for statistical analysis and model validation.
Characterization of ETR-SLN:
Particle size analysis, Polydispersity Index (PDI) and zeta potential: ETR-SLN preparations were dispersed in distilled water and stirred for 5 min to form dispersion and then PDI and mean particle size was measured by dynamic light scattering using a zeta sizer (Malvern Zetasizer). The surface charge was determined by measuring the zeta potential of ETRSLN. Zeta potential measurements were carried out at 25º with an electric field strength of 23 V/cm[18].
Differential Scanning Calorimetry (DSC): DSC was analyzed for dry samples of the SLNs using DSC 60-Shimadzu. Samples were accurately weighed and placed on the aluminum pan and heated at 25°-300° below nitrogen flow. The flow of heat was measured as a sample temperature and used to study the thermal behavior of nanoparticles[17].
X-ray Powder Diffraction (XRPD): X-ray scattering measurements were obtained by employing X-ray powder diffractometer; generated at 40 kV and current of 30 mA with the diffraction angle (2θ) between 5° and 80° at 25° temperature. XRPD studies were carried out for drug and lipid mixtures, the physical mixture containing drug and excipients, and optimized SLN formulation[19].
Release kinetics: In vitro drug release data obtained from release studies were subjected to modeling to assess the kinetics of drug release. Attempts were made to fit the dissolution data into Higuchi model, Korsmeyer-Peppas model and Hixson-Crowell model to determine the mechanism of drug release. The drug release mechanism is explained in terms of the highest correlation coefficient; the highest values of correlation coefficient suggest the release mechanism[20].
Results and Discussion
Solubility of the drug in lipid is a prerequisite for high drug loading in preparation of SLN[13]. To select suitable lipid for the present study, saturation solubility of ETR was done in various lipids and lipids GEL and COM were selected for preparation of SLN. Good solubility of ETR in GEL (stearoyl macrogol glycerides) is due to high Hydrophilic-Lipophilic balance (HLB) value and amphiphilic property[17] and solubility in COM (glyceryl behenate) is attributed to its chemical structure of lipid[15].
Preparation of SLN by hot homogenization technique followed by probe sonication gave uniform dispersed suspension, which was further processed to get dried ETR-SLN powder. ETR-SLN formulations ET1 to ET8 were evaluated for % Y and % EE and results are tabulated in Table 3. The % EE was found to be in the range of 55.6±1.15 to 89.3±1.29 and % Y in the range of 63 % to 83 %. To understand the effect of independent variables on drug dissolution, formulations were subjected to in vitro release studies in comparison with pure drug. The results are depicted in fig. 1a and fig. 1b and ETR-SLN exhibited sustained drug release.

Fig. 1: In vitro drug release, (a) In vitro drug release of SLN formulation ET3 in comparison with pure drug. 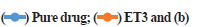 In vitro drug release of SLN formulations ET1 to ET8,
In vitro drug release of SLN formulations ET1 to ET8, 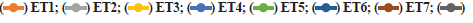 ET8
ET8
Formulations were prepared according to a 23 full factorial experimental design (Table 2) and the observed responses for all formulations are shown in Table 3. Multiple linear regression analysis was done to estimate the effect of factors on responses by generating a polynomial equation:
| Formulation | % Y (Y1) | % EE (Y2) | % DR at 8th h (Y3) |
|---|---|---|---|
| ET1 | 63 | 58.4±1.20 | 21.73±2.90 |
| ET2 | 69 | 55.6 ±1.15 | 22.50±3.44 |
| ET3 | 83 | 87.1±1.57 | 43.68±2.84 |
| ET4 | 75 | 89.3±1.29 | 41.50±3.18 |
| ET5 | 76 | 65.5±1.59 | 17.66±2.82 |
| ET6 | 70 | 81.8 ±1.16 | 30.47±2.8 |
| ET7 | 70 | 63.9±1.41 | 19.70±2.85 |
| ET8 | 73 | 79±1.75 | 43.05±1.57 |
Note: Y1, Y2 and Y3 are responses of SLN formulations
Table 3: In vitro characterizations of SLN formulations
Y=β0+β1A+β2B+β3C+β12AB+β23BC+β13AC+β123ABC
Where Y is the response parameter for each factor level; β0 is an intercept. A, B and C are the coded levels of independent variables. The polynomial equation was used to determine the coefficients and the mathematical sign positive and negative implies a synergistic effect and antagonistic effect. To identify the best fit model, results were subjected to several regression models; Linear, Two-Factor Interactions (2FI) and Three-Factor Interactions (3FI) in the design expert.
Based on the p value, R2 value, predicted R2 value and Predicted Residual Error Sum of Squares (PRESS) value represented in Table 4 and results of Analysis of Variance (ANOVA) represented in Table 5, the best fit significant model for all the responses was found to be linear model represented by the equations as shown in Table 4.
| Reponses | R2 | Adjusted R2 | Predicted R2 | Precision | SD | % CV | p |
|---|---|---|---|---|---|---|---|
| % Yield (Y1) | 0.97 | 0.9534 | 0.8934 | 26 | 1.27 | 1.76 | 0.0013 |
| % Encapsulation (Y2) | 0.99 | 0.9904 | 0.9781 | 26.96 | 1.3 | 1.79 | <0.001 |
| % DR (Y3) | 0.88 | 0.7914 | 0.5232 | 416.22 | 5.1 | 16.98 | 0.0256 |
Note: SD: Standard Deviation; CV: Coefficient of Variation; P: Probability
Table 4: Summary of results of regression analysis for responses Y1, Y2 and Y3
| Parameters | DF | SS | MS | F |
|---|---|---|---|---|
| % Yield | ||||
| Model | 3 | 237.38 | 79.13 | 48.69, significant |
| Residual | 4 | 6.5 | 1.63 | - |
| Total | 7 | 243.83 | - | - |
| % Encapsulation | ||||
| Model | 3 | 1224.73 | 408.24 | 242.28, significant |
| Residual | 4 | 6.74 | 1.69 | - |
| Total | 7 | 1231.74 | - | - |
| % DR | ||||
| Model | 3 | 768.86 | 256.29 | 9.85, significant |
| Residual | 4 | 104.05 | 26.01 | - |
| Total | 7 | 872.91 | - | - |
Note: DF: Degrees of Freedom; SS: Sum of Square; MS: Mean Sum of Square and F: Fischer’s ratio
Table 5: Results of anova for measured responses
As shown in Table 4, the polynomial linear equation generated for responses are as follows.
Y1=72.38+2.87A+3.62B+2.87C,
Y2=72.58+11.72A+3.87B-0.77C,
Y3=30.04+9.64A+0.59B+1.69C
From the generated linear equation and its coefficient values, it is observed that there is no interaction effect (ABC) on the responses. The effect of independent variables A and B shows a positive significant effect on all the response variables Y1, Y2 and Y3. This indicates that the lipid ratio and surfactant concentration has a significant effect on % Y, % EE and % DR. But variable C has a positive effect on % Y and % DR and a negative effect on % EE. For a better understanding of the effect of independent variables on dependent variables and the significance of the effect, surface response plots and Pareto charts were generated for all the responses. Surface response plots and Pareto charts are represented in fig. 2 and fig. 3 for all the responses. During this study, the volume of the continuous phase and the processing variables such as stirring speed and time is kept constant. From fig. 2a it is observed that all three independent factors are responsible for an increase in the % Y, which is significant according to Pareto charts. This indicates that % Y is affected by the concentration of lipids, the ratio of COM:GEL mixture and sonication time.

Fig. 2: Response surface plot, (a) Effect of factors on % yield (Y1); (b) Effect of factors on EE (Y2) and (c) Effect of factors on drug release (Y3) responses

Fig. 3: Pareto chart, (a) Effect of factors on % yield (Y1); (b) Effect of factors on EE (Y2) and (c) Effect of factors on drug release (Y3) responses, where A: Lipid ratio; B: Surfactant concentration and C: Sonication time;  effect.
effect.
Fig. 2b and fig. 3b represents the response surface plot and Pareto chart of % EE. Lipid type and its concentration affect drug % EE. In the present study, it is observed that an increase in lipid ratio (COM:GEL) and surfactant concentration (poloxamer-188) increases % EE. This may be due to high lipid concentration containing a mixture of mono, di and triglyceride which tend to form less perfect crystals which result in greater drug loading capacity[13] and in the presence of poloxamer 188 rapid solubility of the drug occurs in lipid phase enhancing drug encapsulation[8]. However, at the same time, it was observed that an increase in surfactant concentration also resulted in decreased % EE, which may be due to increased solubility of the drug in the aqueous phase during the preparation of SLN in the presence of surfactant[21].
Two responses were observed for the effect of factors on % DR, sustained release of the drug and increase in drug dissolution. Sustained release is attributed to the formation of rigid solid particles due to increased lipid concentration and increased viscosity of the medium and aggregation of particles which occurs at higher sonication speed. Both factors result in decreased drug diffusion to the dissolution medium[22]. Another effect observed was change in the lipid ratio which increases drug release. This effect was due to the lipid GEL, which has the properties of reducing interfacial tension and enhancing the solubilization of poorly soluble drugs[17].
For generating optimized formulation numerical optimization was used by stating the range for independent and dependent variables[23]. Formulation with greater % Y, % EE and % DR was selected as optimized formulation, therefore ET3 with % Y 87 %, % EE 87.1 % and % DR 43.68 % was selected as an optimum formulation with a desirable value of 1. Observed values and predicted values of ET3 were in close relation with predicted values of formulation ET3. The model was validated by preparing ET3 formulation in triplicate and evaluating the response. Predicted average experimental values of ET3 were subjected to conformation study and observed that the average data of the experimentation is within the confirmation view indicating the validity of the model (Table 6). From this result, it indicates that generated linear model describes the effect of independent variables on dependent variables and therefore an optimization technique is an appropriate tool for optimization of ETR-SLN.
| Run 3 response | Predicted mean | Predicted median | Observed | SD | n | SE pred | 95 % PI low | Average data of formulation (ET3) | 95 % PI high |
|---|---|---|---|---|---|---|---|---|---|
| Yield | 81.75 | 81.75 | 83 | 1.2747 | 3.00 | 1.1636 | 78.5191 | 81.4967 | 84.9809 |
| Encapsulation | 87.4 | 87.4 | 87.1 | 1.298 | 3.0 | 1.1849 | 84.11 | 84.3967 | 90.69 |
| Drug release | 41.96 | 41.96 | 43.68 | 5.100 | 3.0 | 4.6559 | 29.033 | 42.2667 | 54.887 |
Note: SD: Standard Deviation; SE: Standard Error and PI: Prediction Interval
Table 6: Confirmation table indicating the validity of the model
Particles with submicron sizes, specifically smaller than 400 nm, may increase the bioavailability of drugs due to decreased particle size[24]. In the present study size of the particle for optimum ET3 was found to be 178.8 nm as shown in fig. 4. This result shows that though a high amount of lipid COM is used, the presence of non-ionic hydrophilic surfactant stabilizes particles in the nano size range[25]. PDI of dispersed particles was found to be 0.075 as shown in fig. 4. PDI is a sign of measurement of the particle size distribution; its value specifies the quality of dispersion. PDI values≤0.1 indicate the highest quality of dispersion[26] and in the present study, the PDI value was observed to be 0.075 indicating a homogenous distribution of SLNs. An increase in concentration of surfactant was found to decrease the PDI value. Homogenization facilitates the particle partition and decreases surface area with an increase in surfactant concentration[27]. Positive or negative zeta potential value signifies more repellant forces and repulsion among the particles with like electrical charges and avoids aggregations of particles and therefore, shows a simple distribution[28]. Zeta potential was measured for ET3 formulation and found to be -36.2 mV as shown in fig. 5, which confirms good physical stability.

Fig. 4: Size distribution of SLN formulation ET3

Fig. 5: Zeta potential of SLN formulation ET3
DSC is used to detect the possible interactions among the components. DSC spectra of drug lipid mixture, physical mixture and optimized preparation of SLN are shown in fig. 6a-fig. 6d.

Fig. 6: DSC endothermic peaks, (a) Physical mixture of ETR:GEL; (b) Physical mixture of ETR:COM; (c) Physical mixture of ETR:GEL:COM and (d) ET3 SLN formulation
The sharp endothermic peaks in fig. 6a and fig. 6b at 280°, 75.01° and 50.93° indicate endotherm of ETR, COM and GEL, and the absence of any interaction between drug and lipids. A decrease in intensity of peak or absence of endothermic peak within the melting point range of ETR in the physical mixture and ET3 preparation confirms the conversion of the crystalline state to an amorphous state of ETR. Literature supports such conversion of drugs from crystalline to amorphous when GEL acts as a carrier in SLN dispersion[13,29].
For the assessment of crystallinity of lipid matrices, XRPD was carried out for drug lipid mixture, physical mixture and SLN formulation ET3, and results are depicted in fig. 7a-fig. 7d. As depicted in fig. 7a sharp peaks in drug lipid mixture at about 2θ-scattered angles indicated the crystalline nature of ETR and absence of interaction. In the XRPD pattern of the physical mixture and ET3 SLN fig. 7d, it was observed that reduced intense peaks of ETR present at the same position, which indicated the absence of interaction and ETR was not in crystalline form in SLN. The amorphous state of ETR in the lipid matrix is the probable reason for the better solubility and dissolution of ETR. These results confirmed that ETR existed in an amorphous state in the SLN formulation[27,30].

Fig. 7: XRD pattern, (a) Physical mixture of ETR:GEL; (b) Physical mixture of ETR:COM; (c) Physical mixture of ETR:GEL:COM and (d) ET3 SLN formulation
To study the release kinetics, data obtained from in vitro drug release studies were subjected to various kinetic models. It was found that the in vitro drug release kinetics of formulation ET3 was best explained by first-order kinetics with the highest linear correlation coefficient (r2=0.9704) followed by zeroorder (r2=0.9708), Higuchi (r2=0.909) respectively as shown in Table 7. From Korsmeyer’s plots, n value was observed to be 0.6219, indicating drug release followed by Non-Fickian diffusion[24,31].
In conclusion, ETR loaded SLNs were successfully prepared by a hot homogenization method followed by probe sonication employing 23 full factorial design. From optimization, it was observed that drug release, % encapsulation and % yield are significantly affected by lipid ratio, surfactant concentration and sonication time. The increase in drug release is attributed to nano sized particles and the presence of GEL and surfactant concentration. Hence prepared nano sized ETR-SLN particles can be effectively targeted to lymphatic circulation for the destruction of the viral reservoir. Further, in vivo studies are required to confirm the benefits of ETR-SLN particles in the management of HIV infection.
Acknowledgements:
We thank Karnataka Lingayat Education (KLE) College of Pharmacy, Bengaluru for providing facilities to carry out this research work.
Conflict of interests:
The authors declared no conflict of interest.
References
- Suri SS, Fenniri H, Singh B. Nanotechnology-based drug delivery systems. J Occup Med Toxicol 2007;2(1):1-6.
[Crossref] [Google Scholar] [PubMed]
- Dizaj SM, Vazifehasl Z, Salatin S, Adibkia K, Javadzadeh Y. Nanosizing of drugs: Effect on dissolution rate. Res Pharm Sci 2015;10(2):95-108.
[Crossref] [Google Scholar] [PubMed]
- Kalepu S, Manthina M, Padavala V. Oral lipid-based drug delivery systems?an overview. Acta Pharm Sin B 2013;3(6):361-72.
[Crossref] [Google Scholar] [PubMed]
- Das S, Chaudhury A. Recent advances in lipid nanoparticle formulations with solid matrix for oral drug delivery. AAPS PharmSciTech 2011;12(1):62-76.
[Crossref] [Google Scholar] [PubMed]
- Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery-A review of the state of the art. Eur J Pharm Biopharm 2000;50(1):161-77.
- Ghasemiyeh P, Mohammadi-Samani S. Solid lipid nanoparticles and nanostructured lipid carriers as novel drug delivery systems: Applications, advantages and disadvantages. Res Pharm Sci 2018;13(4):288-303.
[Crossref] [Google Scholar] [PubMed]
- Bhalekar MR, Pokharkar V, Madgulkar A, Patil N, Patil N. Preparation and evaluation of miconazole nitrate-loaded solid lipid nanoparticles for topical delivery. AAPS PharmSciTech 2009;10(1):289-96.
[Crossref] [Google Scholar] [PubMed]
- Kalam MA, Sultana Y, Ali A, Aqil M, Mishra AK, Chuttani K. Preparation, characterization, and evaluation of gatifloxacin loaded solid lipid nanoparticles as colloidal ocular drug delivery system. J Drug Target 2010;18(3):191-204.
[Crossref] [Google Scholar] [PubMed]
- Havens JP, Podany AT, Scarsi KK, Fletcher CV. Clinical pharmacokinetics and pharmacodynamics of etravirine: An updated review. Clin Pharmacokinet 2020;59(2):137-54.
[Crossref] [Google Scholar] [PubMed]
- Schiller DS, Youssef-Bessler M. Etravirine: A second-generation nonnucleoside reverse transcriptase inhibitor (NNRTI) active against NNRTI-resistant strains of HIV. Clin Ther 2009;31(4):692-704.
[Crossref] [Google Scholar] [PubMed]
- Ojewole E, Mackraj I, Naidoo P, Govender T. Exploring the use of novel drug delivery systems for antiretroviral drugs. Eur J Pharm Biopharm 2008;70(3):697-710.
[Crossref] [Google Scholar] [PubMed]
- Alex MA, Chacko AJ, Jose S, Souto EB. Lopinavir loaded solid lipid nanoparticles (SLN) for intestinal lymphatic targeting. Eur J Pharm Sci 2011;42(1-2):11-8.
[Crossref] [Google Scholar] [PubMed]
- Makwana V, Jain R, Patel K, Nivsarkar M, Joshi A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: Elucidation of mechanism of uptake using chylomicron flow blocking approach. Int J Pharm 2015;495(1):439-46.
[Crossref] [Google Scholar] [PubMed]
- Gupta S, Kesarla R, Chotai N, Misra A, Omri A. Systematic approach for the formulation and optimization of solid lipid nanoparticles of efavirenz by high pressure homogenization using design of experiments for brain targeting and enhanced bioavailability. Biomed Res Int 2017;2017:1-18.
[Crossref] [Google Scholar] [PubMed]
- Aburahma MH, Badr-Eldin SM. Compritol 888 ATO: A multifunctional lipid excipient in drug delivery systems and nanopharmaceuticals. Expert Opin Drug Deliv 2014;11(12):1865-83.
[Crossref] [Google Scholar] [PubMed]
- Hazzah HA, Farid RM, Nasra MM, Hazzah WA, El-Massik MA, Abdallah OY. Gelucire-based nanoparticles for curcumin targeting to oral mucosa: Preparation, characterization, and antimicrobial activity assessment. J Pharm Sci 2015;104(11):3913-24.
[Crossref] [Google Scholar] [PubMed]
- Panigrahi KC, Patra CN, Jena GK, Ghose D, Jena J, Panda SK, et al. Gelucire: A versatile polymer for modified release drug delivery system. Futur J Pharm Sci 2018;4(1):102-8.
- Cometa S, Bonifacio MA, Trapani G, Di Gioia S, Dazzi L, de Giglio E, et al. In vitro investigations on dopamine loaded solid lipid nanoparticles. J Pharm Biomed Anal 2020;185:113257.
[Crossref] [Google Scholar] [PubMed]
- Kelidari HR, Saeedi M, Akbari J, Morteza-Semnani K, Valizadeh H, Maniruzzaman M, et al. Development and optimisation of spironolactone nanoparticles for enhanced dissolution rates and stability. AAPS PharmSciTech 2017;18(5):1469-74.
[Crossref] [Google Scholar] [PubMed]
- Mukherjee S, Ray S, Thakur RS. Solid lipid nanoparticles: A modern formulation approach in drug delivery system. Indian J Pharm Sci 2009;71(4):349-58.
[Crossref] [Google Scholar] [PubMed]
- Derakhshandeh K, Erfan M, Dadashzadeh S. Encapsulation of 9-nitrocamptothecin, a novel anticancer drug, in biodegradable nanoparticles: Factorial design, characterization and release kinetics. Eur J Pharm Biopharm 2007;66(1):34-41.
[Crossref] [Google Scholar] [PubMed]
- Emami J, Mohiti H, Hamishehkar H, Varshosaz J. Formulation and optimization of solid lipid nanoparticle formulation for pulmonary delivery of budesonide using Taguchi and Box-Behnken design. Res Pharm Sci 2015;10(1):17.
[Google Scholar] [PubMed]
- Abbas Z, Marihal S. Gellan gum-based mucoadhesive microspheres of almotriptan for nasal administration: Formulation optimization using factorial design, characterization and in vitro evaluation. J Pharm Bioallied Sci 2014;6(4):267.
[Crossref] [Google Scholar] [PubMed]
- Ebrahimi HA, Javadzadeh Y, Hamidi M, Jalali MB. Repaglinide-loaded solid lipid nanoparticles: Effect of using different surfactants/stabilizers on physicochemical properties of nanoparticles. Daru J Pharm Sci 2015;23(1):1-11.
- Noriega-Peláez EK, Mendoza-Muñoz N, Ganem-Quintanar A, Quintanar-Guerrero D. Optimization of the emulsification and solvent displacement method for the preparation of solid lipid nanoparticles. Drug Dev Ind Pharm 2011;37(2):160-6.
[Crossref] [Google Scholar] [PubMed]
- Shah R, Eldridge D, Palombo E, Harding I. Optimisation and stability assessment of solid lipid nanoparticles using particle size and zeta potential. J Phys Sci 2014;25(1):59-75.
- Hosny KM, Aljaeid BM. Sildenafil citrate as oral solid lipid nanoparticles: A novel formula with higher bioavailability and sustained action for treatment of erectile dysfunction. Expert Opin Drug Deliv 2014;11(7):1015-22.
[Crossref] [Google Scholar] [PubMed]
- Doijad RC, Manvi FV, Godhwani DM, Joseph R, Deshmukh NV. Formulation and targeting efficiency of cisplatin engineered solid lipid nanoparticles. Indian J Pharm Sci 2008;70(2):203-7.
[Crossref] [Google Scholar] [PubMed]
- Sinha S, Ali M, Baboota S, Ahuja A, Kumar A, Ali J. Solid dispersion as an approach for bioavailability enhancement of poorly water-soluble drug ritonavir. AAPS PharmSciTech 2010;11(2):518-27.
[Crossref] [Google Scholar] [PubMed]
- Ramalingam P, Ko YT. Improved oral delivery of resveratrol from N-trimethyl chitosan-g-palmitic acid surface-modified solid lipid nanoparticles. Colloids Surf B Biointerfaces 2016;139:52-61.
[Crossref] [Google Scholar] [PubMed]
- Merchant HA, Shoaib HM, Tazeen J, Yousuf RI. Once-daily tablet formulation and in vitro release evaluation of cefpodoxime using hydroxypropyl methylcellulose: A technical note. AAPS PharmSciTech 2006;7(3):178-83.
[Crossref] [Google Scholar] [PubMed]





