- *Corresponding Author:
- M. V. Ramana
Professor, Pharmaceutical Chemistry, School of Science and Humanities, VIT University, Vellore - 632 014
E-mail: dr_ramanamv@yahoo.com
| Date of Submission | 14 October 2005 |
| Date of Revision | 21 February 2007 |
| Date of Acceptance | 21 May 2007 |
| Indian J Pharm Sci, 2007, 69 (3):390-393 |
Abstract
Liposomal encapsulation is known to significantly improve the therapeutic index of a drug. In the present investigation liposomal formulations were chosen to transport clindamycin, which is considered as the most effective topical antibiotic for acne, into the skin layers. Liposomes with clindamycin phosphate were prepared using lipid film hydration method and the optimum ratio of the components was determined. The liposomes were characterized for their vesicle size, shape, encapsulation efficiency, % drug content and for in vitro skin permeation study. The results suggest that the average size of vesicles was found to be in range of 4.91-6.75 µm. Highest encapsulation efficiency (45.4%) and in vitro skin permeation (62%) was achieved with a formulation containing drug: lipid: cholesterol in the ratio of 1:1:1. Liposomal formulation of clindamycin phosphate with good skin permeation properties was incorporated into gel base and comparison of in vitro skin permeation was made with non liposomal marketed gel, both containing 1% clindamycin phosphate. Higher rate of drug release across the rat abdominal skin was found with liposomal gel (54%) than non-liposomal marketed gel (48.7%). Biological study revealed that by encapsulating clindamycin phosphate into liposomes the occurrence of Pseudomembranous colitis could be reduced significantly in comparison to plain clindamycin phosphate.
Keywords
Liposomes, clindamycin, Pseudomembranous colitis, Clostridim difficile, antidiarrheal activity
Liposomes are minute spherical vesicles consisting of lipid bilayers and have been suggested for use as a suitable carrier for various drugs used in oral, topical and parenteral administration [1-5]. Liposomal drug formulations are able to reduce side effects and incompatibilities to enhance the accumulation of the drug at the administration site and to incorporate a variety of hydrophilic and hydrophobic drugs [3]. Clindamycin is generally considered the most effective topical antibiotic for acne. Clindamycin is reported to produce diarrhea in about 20% of patients after systemic administration; in some patients severe antibiotic-associated Pseudomembranous colitis, which sometimes, a potentially a lethal condition, may develop [5,6]. This is due to an acute inflammation of colon caused by a necrotising toxin produced by clindamycin resistant organism, Clostridium difficile, which may be part of normal faecal flora [6,7].
The aim of the present investigation was to prepare clindamycin phosphate liposomal systems and to compare their topical performance with non liposomal marketed system containing clindamycin phosphate by determining permeability of drug through the skin layer. The purpose of the present work was also to investigate the effectiveness of liposomal formulation of clindamycin phosphate in reduction of Pseudomembranous colitis in comparison to plain drug formulation after oral administration.
Materials and Methods
Clindamycin phosphate (CMP) was obtained from Group Pharmaceutical limited, Bangalore as gift sample; soya lecithin and cholesterol were purchased from Ranbaxy Fine Chemicals Ltd., Delhi. Micrococcus luteus (ATCC 9341) was obtained from NCIM, Pune. All other reagents used were of analytical reagent grade.
Preparation of liposomes
Liposomes containing CMP were prepared by thin film hydration method [8]. Ten ml chloroform solution containing specified quantities of soya lecithin and cholesterol (Table 1) were added into a round bottom flask. The organic solvent was removed by using a rotary flash evaporator at 40° by applying vacuum. This results in the formation of a thin film in the flask. A solution of CMP in 10 ml of 0.05 M phosphate buffer pH 6.0 was added drop wise into the flask, while agitating the flask vigorously using a vibrator (Wiswo Instruments, Mumbai). The resultant suspension was sonicated using a probe sonicator (Bandelin Sonopuls, HD2070). Five formulations (F1 to F5) containing different ratio of drug, lipid and cholesterol were prepared and characterised with respect to size distribution and average vesicle size, shape, % drug content, encapsulation efficiency and in vitro skin permeation.
| Formulation | Formulation ratio | ||
|---|---|---|---|
| Drug | Lipid | Cholesterol | |
| F1 | 1 | 1 | 1 |
| F2 | 1 | 2 | 1 |
| F3 | 1 | 1 | 2 |
| F4 | 2 | 2 | 1 |
| F5 | 2 | 1 | 2 |
Table 1: Composition of Prepared Liposomes
Microscopy
All the batches of the liposomes prepared were viewed under binocular compound microscope (Nikon), to study their size and shape. Size of 150 liposomal vesicles from each batch was measured at different locations of slide and average size of liposomal vesicles was determined. The results are given in Table 2 and photomicrograph is given in fig. 1.
| Formulation | Average size (µm)* | % Encapsulationefficiency* | Total % drugcontent | % drugpermeationafter 24 h |
|---|---|---|---|---|
| F1 | 4.91±0.81 | 45.4 | 99.2±1.23 | 62.8 |
| F2 | 5.20±0.916 | 39.8 | 98.1±1.34 | 60.2 |
| F3 | 6.75±0.535 | 33.7 | 95.4±2.12 | 53.0 |
| F4 | 5.79±1.02 | 27.5 | 95.1±1.58 | 54.9 |
| F5 | 6.25±1.07 | 23.8 | 94.6±2.34 | 49.0 |
*Average of 3 determinations
Table 2: Characterization of Differentliposomal Formulations

Figure 1: Liposomal formulation (F1) under 45X 10 magnification
Encapsulation efficiency and % drug content
The liposomal suspension was centrifuged for 1 h at 20,000 rpm to allow the sedimentation of liposomes in the form of pellet and clear supernatant was decanted.
The amount of CMP in supernatant was determined by microbiological method using a standard graph prepared for CMP by microbiological assay method [9] using Micrococcus luteus (ATCC 9341). Residue was lysed using 1% v/v solution of triton-X-1003 (0.2 ml in 2 ml liposomal suspension) by vortexing and then centrifuging it for 1 h at 20,000 rpm. The amount of drug entrapped was determined by microbiological method using the standard graph. Percentage drug content was calculated by adding the amount of drug present in supernatant and amount of drug entrapped in vesicles. The results are given in Table 2.
In vitro skin permeation study
In vitro skin permeation study was carried out for the formulations F1 to F5 using a modified Keshary- Chein diffusion cell. Rat abdominal skin was used to determine drug permeability [2,10]. Twenty five millilitres of phosphate buffer saline (pH 6.4) at 37° was used as receptor fluid. Two millilitres of each sample was added to donor compartment. Samples (1 ml each) were withdrawn periodically from receptor fluid and the drug content was determined by microbiological method using a standard graph. The results are given in Table 2 and fig. 2.

Figure 2: In vitro skin permeation study for liposomal formulation F1, F2, F3, F4 and F5
In vitro skin permeation of liposomal formulations F1 (─ ─), F2 (─■─), F3 (─▲─), F4 (─•─) and F5 (─
─), F2 (─■─), F3 (─▲─), F4 (─•─) and F5 (─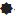 ─)
─)
Preparation of topical dosage form
The drug-loaded liposomes, after separation of free drug, were incorporated into carbopol 940 gel base [11] in such a way that final formulation contains 1% w/w of CMP. In vitro skin permeation study of formulated liposomal gel and marketed non-liposomal gel both containing 1% w/w CMP was carried out using a rat abdominal skin in a similar way as described under ‘in vitro skin permeation study’. The results are given in Table 3 and fig. 3.
| Formulation | Cumulative % drug permeated after 24 h |
|---|---|
| Formulated liposomal gel | 54.0 |
| Marketed non-liposomal gel | 48.7 |
Table 3: Cumulative Amount of Drug Permeated After 24 H From Different Formulations

Figure 3: In vitro drug release study for formulated liposomal gel and
marketed non-liposomal gel
In vitro drug release of formulated liposomal gel (--) and
marketed non-liposomal gel (-•-)
Preparation of oral dosage form
A suspension was prepared for oral administration after separation of free drug from liposomal entrapped drug. These separated liposomes containing 100 mg of CMP in entrapped form were diluted to 30 ml with 0.05 M phosphate buffer (pH 6.0) containing 0.5% carbopol 940 as suspending agent11. Similarly, oral solution containing pure CMP was prepared in the same concentration as oral liposomal suspension of CMP.
Biological studies for a possible reduction in diarrhea using liposomal suspension of CMP
Animal experiments were carried out according to CPCSEA and institutional ethical committee guidelines. The ethical clearance certificate was obtained from the institutional ethical committee. Wistar rats of either sex weighing between 260-310 g were used in the test. The extent of induction of diarrhea (Pseudomembranous colitis) in experimental animals, when clindamycin formulations were administered, was determined. The animals were administered with different formulations such as suspension of pure CMP, liposomal suspension of CMP, and drug free plain liposomal suspension (control). The animals were divided into 3 groups of 20 animals each and were allowed free access to food and water. The animals were maintained on normal diet, water was made available all the time and rats were kept in normal temperature and humidity condition. Calculated volume of each formulation based on the body weight of each animal (30 mg/kg body weight, bid) was administered by oral route with oral feeding tube for seven days [4]. Number of animals that developed symptoms of diarrhea were counted in each group and separated from other animals in the group. Fecal matter expelled by diarrhea induced animals and rest of the animals in the group was collected separately at 12 h interval up to 10 d and weighed; fecal matter was dried at 50° for 8 h in an oven and weighed again. Fluid content in the fecal matter was determined by calculating the difference in weight between the initial weight and weight after drying [12]. The results are given in Table 4.
| Formulation treatment | Dose(mg/kg/d) | Animalsused (n) | Animalsshowingdiarrhea (n) | Average % fluid content± SE |
|---|---|---|---|---|
| Pure clindamycinsolution | 30 | 20 | 4 | 85.86±0.944 |
| Liposomalclindamycin suspension | 30 | 20 | 1 | 64.50±0.787 |
| Plain liposomalsuspension (control) | - | 20 | 0 | 60.4±0.513 |
Table 4: Data Showing Average % Fluid Content In All Three Groups
Results and Discussion
Liposomal system of different compositions was prepared and characterised (Tables 1 and 2). The average size of liposomes was found to be in the range of 4.91-6.75 μm. Shape of most of CMP containing liposomes was found to be spherical (fig. 1). Entrapment efficiency also showed that a significant amount of CMP was entrapped in liposomal vesicles. The entrapment efficiency was found to be maximum with formulation containing soya lecithin and cholesterol in a ratio of 1:1 (F1). Increasing the concentration of cholesterol increased lipophilic properties of lipid bilayer hence decreased entrapment of hydrophilic drug like CMP. In liposomes with less amount of cholesterol, the lipid bilayer vesicles become less stable and delicate which leads to a rupture of the vesicles and decrease in entrapment efficiency because of leaking of drug from the vesicles. Hence a balanced concentration of cholesterol is essential to form stable liposomes. The total % drug content in the formulation range from 94.6-99.2% indicates that there is no degradation of drug in the process. In vitro skin permeation study for all five formulations was found that formulation F1 showed higher permeation rate than other formulations. This is due to smaller size of the vesicles (4.91 μm) and higher drug entrapment (45.5%) in the liposomal vesicles. In vitro skin permeation studies of liposomal formulations have shown significant permeation of the drug (49.0%-62.8%). Permeation of drug can further be enhanced by incorporating additives such as permeation enhancers or using similar techniques to enhance the permeation of drugs.
On the basis of physical characteristics, drug entrapment efficiency and in vitro skin permeation studies the liposomal formulation F1 was selected for its incorporation into gel base. The formulated liposomal gel and marketed non-liposomal gel, both containing 1% CMP were subjected to in vitro skin permeation studies across rat abdominal skin and comparison of results was done. It was found that liposomal gel showed higher permeation (48.7%) than non-liposomal gel (43.0%) at the end of 24 h study. Phospholipids, being amphiphilic in nature can modify the properties of the keratinized layer of the skin and enhance the permeation of drug through the skin.
Biological activity for a possible reduction in diarrhea was carried out in albino rats and results obtained reveal that in 4 out of 20 animals administered with suspension of pure drug have developed the symptoms of diarrhea in comparison to one out of 20 animals administered with liposomal suspension of CMP. The group treated with pure CMP solution showed higher percentage fluid content in faeces than that were treated with liposomal suspension of CMP. These results indicated that the occurrence of diarrhea (Pseudomembranous colitis) due to CMP is reduced significantly by the administration of the drug in the form of liposomal suspension. This is due to small size of liposomes, which can be absorbed rapidly from the GI tract in intact form thus reducing the contact of drug with gastrointestinal tract. Thereby normal flora of intestine is protected and colonisation of Clostridium difficile is prevented.
Hence it is concluded that administration of CMP form of liposomes can reduce the occurrence of Pseudomembranous colitis and the topical formulation of liposomes containing CMP showed much greater permeability of drug into skin layers compared to non-liposomal marketed formulation.
Acknowledgements
The authors are extremely grateful to Group Pharmaceuticals, Bangalore for providing gift sample of clindamycin phosphate.
References
- Sood A, Venugopalan P, Venkatesan N, Vyas SP. Liposomes in cosmetics and skin care. Indian Drugs 1996;33:43-9.
- Pillai O, Panchagnula R. Transdermal iontophoresis of insulin VI: Influence of pretreatment with fatty acids on permeation across rat skin.Skin Pharmacol Physiol 2004;17:289-97.
- Singh R, Vyas SP. Topical liposomes for localized and controlled drug delivery. J Dermatol Sci 1996;13:107-11.
- Sanic A, Kilinc M, Erdogan S, Ozer AY, Durupinar B. Comparison of the in vitro efficacies of liposomal vancomycin with Vancomycin and liposomal ofloxacin with ofloxacin against Staphylococcus aureus. Clin Microbiol Infect 2001;7:1-394.
- Patel HM, Ryman BE. Oral administration of insulin by encapsulation within liposomes. FEBS Lett 1976;62:60-3.
- Kelly CP, Lamont JT. Clostridium difficile infection. Annu Rev Med 1998;49:375-90.
- Parfitt K, editor. In: Martindale, the complete drug reference. 32nd ed, Pharmaceutical Press: London; 1999, 191.
- Szoka F, Papahadjopoulos D. Comparative properties and methods of preparation of lipid vesicles (liposomes). Ann Rev Biophys Bioeng 1980;9:467-508.
- Indian Pharmacopoeia, Vol. II, Govt. of India, Ministry of Health and Family Welfare, Delhi; The Controller of Publications, 1996. p. A-100-8.
- Ferreira LS, Ramaldes GA, Nunan EA, Ferreira LA. In vitro skin permeation and retention of paromomycin from liposomes for topical treatment of the cutaneous leishmaniasis. Drug Dev Ind Pharm 2004;30:289-96.
- Rowe RC, Sheskey PJ, Weller PJ, editors, Handbook of pharmaceutical excipients, 2nd ed. Pharmaceutical Press and American Pharmaceutical Association: 1994. p. 71.
- Vogel HG, editor. Drug discovery and evaluation: Pharmacological assays, 2nd ed. New York; Springer Verlag: 2002. p. 877.





