- *Corresponding Author:
- Ying Cui
Department of Oncology, State Key Laboratory of Targeting Oncology, National Center for International Research of Bio-Targeting Theranostics, Guangxi Key Laboratory of Bio-Targeting Theranostics, Collaborative Innovation Center for Targeting Tumor Diagnosis and Therapy, Guangxi Medical University, Nanning, Guangxi 530021, China
E-mail: Ycuigx@163.com
| This article was originally published in a special issue, “Recent Progression in Pharmacological and Health Sciences” |
| Indian J Pharm Sci 2024:86(2) Spl Issue “210-221” |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
This study aimed to investigate the potential mechanisms underlying the therapeutic effect of matrine in osteosarcoma using network pharmacology and single-cell ribonucleic acid sequencing. Matrine's potential gene targets were sourced from multiple databases; comparative toxicogenomics database, SwissTargetPrediction, Binding DB, and TargetNet. We extracted gene expression data related to osteosarcoma from the GSE14359 dataset. To discern the activation pathways of genes linked to both the drug and disease, we performed differential gene analysis, Kyoto encyclopedia of genes and genomes pathway analysis, and gene ontology enrichment analysis. By employing both weighted gene co-expression network analysis and differential analysis, we pinpointed critical genes associated with osteosarcoma. Protein-protein interaction analysis was utilized to highlight hub genes. Furthermore, module analysis revealed key gene clusters that affect matrine. For a comprehensive evaluation of matrine’s pharmacological targets and signaling pathways in osteosarcoma, single-cell ribonucleic acid sequencing was carried out on the GSE162454 dataset using the Seurat package in R. A total of 15 cell clusters were identified, and the analysis results suggested that the mechanism of matrine's action on osteosarcoma may involve macrophages. This study elucidated the targets and pathways through which matrine acts on osteosarcoma using network pharmacology. Moreover, single-cell sequencing identified macrophages as key cellular markers for matrine's action in osteosarcoma. These findings provide new therapeutic evidence for clinical treatment strategies.
Keywords
Weighted gene co-expression network analysis, osteosarcoma, macrophages, network pharmacology, metastasis
Osteosarcoma (OS) is a primary malignant disease predominantly affecting children and adolescents, frequently associated with a bleak prognosis[1]. Despite extensive research, the fundamental mechanisms driving OS are yet to be fully understood. A key characteristic of OS is its tendency for early metastasis, predominantly to the lungs, which constitutes >85 % of metastatic cases[2]. Lung metastasis stands as the principal cause of mortality in OS patients. The overall survival rate for metastatic OS remains at a disheartening 20 % after 5 y, indicative of the grim prognosis[3,4]. Standard treatment approaches for OS encompass adjuvant chemotherapy and surgical intervention[5]. While a variety of chemotherapeutic agents are utilized in the clinical setting, high-dose chemotherapy often yields severe side effects, and drug resistance is common[6,7]. Therefore, the exploration of novel therapeutic agents to bolster OS treatment efficacy is of paramount importance.
Matrine, a naturally occurring alkaloid from the leguminous plant Sophora flavescens (S. flavescens), has gained considerable interest due to its antiinflammatory and antineoplastic properties[8,9]. In our country, the use of S. flavescens injection as an adjuvant therapy for tumors is common in clinical practice. Matrine displays robust anti-tumor activity; it restricts the proliferation of various tumor cells, induces cell cycle arrest, promotes cell apoptosis, curtails metastasis and invasion, reverses multidrug resistance, and reduces the toxic side effects of chemotherapy and radiotherapy[10-12]. Additionally, matrine demonstrates unique impacts on the proliferation, invasion, and drug resistance of OS cells[13]. Despite its array of biological activities, the bioavailability of matrine remains low, considerably limiting its clinical usage. Moreover, compared to other tumor types, research into the effect of matrine on OS is relatively scarce. Therefore, a comprehensive study of matrine's molecular mechanism in OS using network pharmacology and single-cell Ribonucleic Acid (RNA) sequencing could offer valuable insights for its clinical application and development as a therapeutic drug[14].
In this research, we investigated the mechanism through which matrine acts on OS using a network pharmacology approach. We analyzed transcriptional data and clinical information derived from the Gene Expression Omnibus (GEO) database to identify Differentially Expressed Genes (DEGs). This allowed us to discuss the correlations between the genetic and immune functions of drugs and diseases, pinpointing potential biological targets. Subsequently, we verified our findings using the GEO single-cell dataset, which confirmed that matrine impacts macrophages to control cytokine activity and enhance human immune function. Flowchart was shown in fig. 1.

Fig. 1: Flowchart of the GEO single-cell dataset
Materials and Methods
Matrine target screening:
To pinpoint matrine’s potential targets, we extracted data from four reputable databases; the Comparative Toxicogenomics Database (CTD), BindingDB, TargetNet, and SwissTargetPrediction. We meticulously identified matrine’s target genes from these sources. Upon extraction, we removed duplicates to finalize a list of potential targets. Using the clusterProfiler package in R, we conducted Gene Ontology (GO) functional annotation for these candidates, spanning Molecular Function (MF), Cellular Component (CC), and Biological Process (BP) domains. Furthermore, we undertook the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis and implemented Disease Ontology (DO) functional annotation for these genes. We visualized the outcomes via the ggplot2 tool in R, setting a significance threshold with adjusted p<0.05.
Identification of differential genes in OS:
The GSE14359 dataset was retrieved from the GEO database, encompassing 2 primary osteoblasts, 5 conventional OS samples, and 4 OS lung metastasis specimens. Upon securing the dataset, normalization was executed on these samples. Following this, we employed the ‘limma’ package in R to conduct a differential gene analysis between primary osteoblasts and OS sample controls, setting the screening criteria as [log2 fold-change]>1 and p<0.05. The differential gene expression was subsequently visualized using the 'ggplot2' and 'heatmap' packages in R. We then performed Gene Set Enrichment Analysis (GSEA) on the differential genes, utilizing the 'org. Hs. eg. db' and 'clusterProfiler' packages in R[15]. Post gene enrichment analysis, the gene set size was constrained to be ≥500, yielding the gene enrichment result.
Analysis of GSE14359 using weighted gene coexpression network:
Utilizing the Weighted Gene Co-Expression Network Analysis (WGCNA) package in R, we assess the conformity of representative genes to the scalefree distribution's soft-threshold by evaluating their scale independence (R2) and average connectivity. Subsequently, we utilize the Pearson correlation method to discern modules and their pivotal genes linked to migration. In the process of gene set module clustering, by setting minModuleSize to at least 100 and MEDissThres to no <0.3, cluster analysis is performed on modules, and similar modules are combined into new modules. The WGCNA algorithm in R customarily ascertains the associations between disease subtype phenotypes and module genes, by calculating the interrelations among modules. The intensity of these associations is often depicted via a heatmap. Regarding the filtered gene modules, we classify key genes as those exhibiting a Module Membership (MM) exceeding 0.8 and Gene Significance (GS) surpassing 0.65, forming the basis of the key gene module. Lastly, we discern disease-associated genes by intersecting key module genes with previously chosen differential genes. Subsequently, these disease-related genes are subjected to GO and KEGG pathway enrichment analysis (p<0.05)
Building the Protein-Protein Interaction (PPI) network:
The PPI network is constructed using the search functionality of the STRING database (v11.5). The previously identified drug targets and diseases are imported to construct the PPI network, with the original data saved in Tab-Separated Values (TSV) format. Cytoscape (version 3.9.1) is employed to construct and visualize the PPI network. The MOCODE plug-in (molecular complex detection) is used to isolate the core subnetwork in the PPI network, facilitating a deeper understanding of OS prognosis. Subsequently, this subnetwork is subjected to KEGG enrichment analysis.
Visualization of immune infiltration and differential expression:
Upon identification of the key gene subsets, we employ the 'IOBR' package in R to conduct an immune infiltration analysis. The CIBERSORT bioinformatics algorithm is utilized to estimate the relative proportions of cell types via gene expression data maps, and vector regression methods are employed to process gene expression data of the GSE14359 dataset, aiding in the estimation of the proportions of diverse cell types[16]. We also scrutinize the differential expression of key gene subgroups between the disease and control groups (p<0.05 and absolute log2 fold-change >1), leveraging R for result visualization.
Single-cell RNA sequencing for OS-related gene identification:
Single-cell sequencing analysis was performed on the public dataset GSE162454, which contains biopsies from six OS patients, consolidated into three cases. Raw data was preprocessed utilizing the Seurat R package, wherein single-cell genes were filtered to maintain a count between 300 and 5000; cells with mitochondrial content exceeding 10 % were considered as low-quality and subsequently filtered out. We then employ the Harmony package to mitigate batch effects in the cell data. The filtered primary cell components are clustered, and projection dimensionality reduction technology is applied for visual classification. The identified key gene subgroups associated with matrine are projected onto the cell clusters, yielding potential matrine targets for OS treatment.
Results and Discussion
Following the examination of four databases to identify the drug targets of matrine, we retrieved a total of 194 matrine-associated target genes (fig. 2A). We conducted GO, DO, and KEGG enrichment analysis, revealing that GO, BP were predominantly enriched in response to xenobiotic stimulus, cellular response to monoamine stimulus, and cellular response to catecholamine stimulus, among others. The GO, CC were primarily enriched in synaptic regions and neuronal cells, among others. These results suggest that matrine may influence the nervous system, impacting the transmission of nerve signals and the function or survival of nerve cells. GO, MF were predominantly enriched in neurotransmitter receptor activity, G proteincoupled amine receptor activity, and serotonin receptor activity (fig. 2B). DO enrichment primarily included hepatitis, myocardial infarction, and musculoskeletal system cancer (fig. 2C and fig. 2D), among others. Importantly, musculoskeletal system cancer encompasses OS, implying that matrine's potential targets might be influential in OS. KEGG enrichment analysis indicated a primary enrichment in neuroactive ligand-receptor interaction, pathways of neurodegeneration-multiple diseases, cyclic Adenosine 3′,5′-Monophosphate (cAMP) signaling pathway, and the Phosphatidylinositol-3-Kinase (PI3K)-Protein Kinase B (Akt) signaling pathway (fig. 2E and fig. 2F).

Fig. 2: Matrine targets, (A): Venn diagram of matrine targets in four database; (B): GO enrichment analysis of matrine targets; (C and D): DO
enrichment analysis of matrine targets and (E and F): KEGG matrine target enrichment analysis Note: 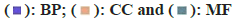
After normalizing the GSE14359 data set (fig. 3A and fig. 3B), we screened a total of 1805 DEGs between OS and normal samples, including 877 up-regulated genes and 928 down-regulated genes. Visual representations were constructed by creating volcano and ridge plots using the R programming language (fig. 3C and fig. 3D). Subsequently, we performed GSEA pathway enrichment using OS samples and normal samples. Results revealed enrichment of multiple pathways in OS, including allograft rejection, asthma, autoimmune thyroid disease, primary immunodeficiency, and systemic lupus erythematosus. These findings suggest that immune dysfunction plays an important role in the development of OS.

Fig. 3: DEGs in GSE14359, (A): Dataset before and after normalization; (B): GSEA analysis base on KEGG analysis, (C): Volcano map of dataset
DEGs and (D): Ridge plot showing DEGs enrich pathways in GSEA Note: 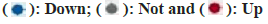
We downloaded the GSE14359 dataset for further analysis and proceeded with processing the abnormal data. Next, we analyzed the gene reads module and selected those genes with Fragments Per Kilobase Million (FPKM) exceeding 0.5 for further investigation. We set a soft threshold range from 1-30, ensuring an R2 value (a measure of certainty) of >0.9. Further WGCNA analysis reveals that upon finding the intergenic soft threshold to be 7 (fig. 4A) by measuring independence and average correlation comparison, we utilized the topological overlap matrix to construct a hierarchical tree among genes. Consequently, 26 modules were generated by merging similar modules (fig. 4B) among with, the DarkOliveGreen and MediumPurple3 modules exhibited high correlation (r>0.8, p<0.001). Scatterplots demonstrated a pronounced correlation between GS and MM in both modules (fig. 4C and fig. 4D). Consequently, the DarkOliveGreen and MediumPurple3 modules may represent key modules in OS.

Fig. 4: WGCNA, (A): Selection of soft threshold; (B): Correlation of module eigengenes with osteosarcoma and (C and D): Correlation of DarkOliveGreen and MediumPurple3 with osteosarcoma
We compared genes from two modules with the DEGs from GSE14359, as illustrated in fig. 5A. Subsequently, a comprehensive analysis was conducted on the overlapping genes using both GO and KEGG enrichment methodologies. The GO enrichment analysis predominantly underscores pathways, including but not limited to, positive regulation of cell activation, cell chemotaxis, outer plasma membrane, endocytic vesicles, kinase regulatory activity, and integrin binding. In contrast, the KEGG enrichment analysis elucidates a marked enrichment in pathways such as the Mitogen- Activated Protein Kinase (MAPK) signaling pathway, PI3K-Akt signaling pathway, Rap1 signaling pathway, and the Ras signaling pathway, as shown in fig. 5B-fig. 5F.

Fig. 5: Targets, (A): Venn diagram of WGCNA and differential genes and (B-F): GO and KEGG analysis of disease genes
Disease-enriched genes and molecular drug targets were imported into the STRING online database (v11.5) (https://www.string-db.org/) to facilitate the construction of the PPI network (fig. 6A). The correlation degree was established at 0.70, and unconnected nodes were excluded, culminating in the formation of a protein interaction network. Utilizing Cytoscape's MOCODE, 25 essential subgroup genes were identified (score=17.667) (fig. 6B). The 25 identified genes underwent DO and KEGG analysis, revealing the main enrichment results of DO; cell type benign neoplasm, ovarian cancer, and musculoskeletal system cancer, among others. These results included human cytomegalovirus infection, lipid and atherosclerosis, and the PI3K-Akt signaling pathway (fig. 6C and fig. 6D). An intersection between the prior KEGG and DO analysis revealed the recurrence of both musculoskeletal system cancer and the PI3K-Akt signaling pathway, suggesting a strong correlation between matrine and OS, potentially mediated through the PI3K-Akt signaling pathway. Apart from modulating cell metabolism pathways, matrine also influences OS growth by curbing tumor angiogenesis.

Fig. 6: Analysis of key genes, (A and B): PPI network and gene groups and (C and D): Enrichment analysis of essential
sub-cluster gene of Kyoto
Note: 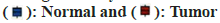
Fig. 7A illustrates significant disparities in key gene clusters between the disease and control groups. Analysis via CIBERSORT revealed that major regulatory targets of the key gene clusters were predominantly within immune pathways, including macrophages_M0, macrophages_M2, and T cells CD4 memory resting (fig. 7B-fig. 7D). This implicates these genes in the regulation of immune function in OS.

Fig. 7: Relationship between key cluster genes and immune infiltration, (A): Differential expression of crucial subgroup genes in tumor and control tissues; (B): Schematic diagram of CIBERSORT and (C and D): Relationship between key gene clusters and immune infiltration
Analysis of OS samples revealed a significant positive correlation between the quantified gene expression and the number of genes detected in the cells. However, mitochondrial expression seemed to operate independently of the general gene expression. To ensure the integrity of our cell analysis, cells exhibiting over 10 % mitochondrial expression were excluded. Our quality control and screening methodologies for the single-cell sequencing of OS samples are illustrated in fig. 8A and fig. 8B. We employed principal component analysis for clustering, setting a resolution of 0.5 (fig. 8C-fig. 8E). The heat map in fig. 9A showcases different genotypes, while the unified manifold approximation and projection in fig. 9B differentiate 15 unique cell clusters, each delineated by distinct colors. Further, the distribution of the AU cell functional score in various cells indicates that matrine predominantly impacts macrophages, as seen in fig. 9C and fig. 9D.

Fig. 8: Comparison of single cell analysis before and after standardization, (A and B): Quality control analysis of single cell datasets; (C and D):
Principal component analysis (PCA) plots before and after and (E): PCA heat map Note: 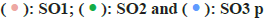

Fig. 9: Matrine pathways of action, (A): Unified manifold approximation and projection clustering into 15 clusters; (B and C): Matrine drugs pathways of action and (D): Cell marker heat map
OS, a prevalent primary malignant tumor, is most commonly observed in adolescents and presents with a poor prognosis and high mortality rate[17]. It encompasses several types based on histopathology, such as the osteoblastic type and chondrogenic type, with survival prognoses showing no notable differences among these types[18]. Despite chemotherapy and surgery being the primary treatment options, their efficacy in treating malignant OS is subpar. Approximately 68 % of localized OS patients survive for 5 y or longer[19]. Over time, due to repeated and long-term chemotherapy, tumor cells may develop a decreased sensitivity or resistance to drugs[20]. This resistance culminates in chemotherapy failure, adversely impacting the treatment outcomes and survival prognoses for OS patients.
Matrine, a principal component of the traditional Chinese medicine S. flavescens, displays potent anti-tumor properties. It is known to be effective against various types of cancer such as liver cancer, colorectal cancer, and lung cancer[21-23]. Recently, the effectiveness of matrine against OS has been substantiated through experimentation[24]. Our experiment primarily involves utilizing single-cell sequencing for comprehensive investigation and genomic-level analysis. This method is instrumental in highlighting tumor heterogeneity, thus playing a significant role in evaluating the treatment outcomes and survival prognoses of OS. Furthermore, singlecell sequencing significantly impacts network pharmacology[24]. It aids in precision targeting of drugs, evaluating drug efficacy, and providing a better understanding of the molecular mechanisms underlying drug action.
Utilizing a collection of databases, we identified target genes associated with matrine. GO enrichment results reveal that these genes are primarily enriched within synaptic transmission regions and neuronal cells when observing CC. BP primarily see enrichment in responses to exogenous stimuli, cellular reactions to monoamine stimuli, and cellular responses to catecholamine stimuli. In the domain of MF, enrichment is mainly observed in neurotransmitter receptor activity. Disease enrichment demonstrated that targets were largely found in cancers of the musculoskeletal system, obstructive lung disease, and disorders of the female reproductive system. Furthermore, KEGG enrichment indicates a concentration of primary targets in pathways such as the PI3K-Akt signaling and cAMP signaling. These findings suggest matrine interacts with various tumor cells by acting upon transmitter cells and other signaling pathways. Following this, we gathered DEGs of OS, which included 877 up-regulated genes and 928 down-regulated genes. GO enrichment was primarily observed in phagocytosis, positive regulation of cell activation, and endocrine vesicle membrane. The implication of rheumatoid arthritis, Staphylococcus aureus infection, and the PI3K-Akt signaling pathway, alongside phagosome, hints at an activation of the immune system within tumor tissues.
We proceeded to identify the key genes, and our findings indicated a consistent enrichment in the PI3KAkt signaling pathway and musculoskeletal system cancer, corroborating our previous verification[25]. The Cibersort cell fraction demonstrated that the key gene cluster related to drug disease is a primary immune pathway and the target of other pathways, thus playing a therapeutic role. Analysis from singlecell sequencing revealed that matrine's main target was macrophages[26].
Matrine enhances the prognosis of OS by interacting with macrophages and stimulating synaptic transmitter transmission. The consistency of our key gene cluster verification with previous results was also confirmed. Lin et al.[27] proposed that Interleukin-6 (IL-6) enhances migration and Intercellular Adhesion Molecule-1 (ICAM-1) expression in human OS cells, potentially activated via the PI3K-Akt signaling pathway, suggesting a potential target for clinical treatments. Wang et al.[28] discovered that microRNA (miR)-143 suppresses Epidermal Growth Factor Receptor (EGFR) signaling via its downstream Extracellular Signal-Regulated Kinase (ERK)/MAPK signaling cascade, thereby inhibiting OS invasion. Liu et al.[29] demonstrated that knocking down AKT1 considerably curtailed OS cell viability, thereby bolstering the assertion that OS is tightly linked to the dysregulation of the PI3K/AKT signaling pathway. Through miR-134, Chen et al.[30] managed to reduce the expression of MMP1 and MMP3, consequently affecting the migratory ability of OS cells. Zhang et al.[31] through lentivirus-mediated RNA interference, were able to silence the expression of Cyclin D1 (CCND1) and observed that this silencing enhanced the cytotoxicity of Doxorubicin (DXR) in MG63 cells. This provides new insights into potentially more effective therapeutic strategies to overcome chemo resistance in OS. L
i et al.[32] by knocking down the ADP-Ribose Polymerase 1 (PARP-1), demonstrated that PARP-1 plays a pivotal role in the regulation of OS growth. Xu et al.[33] confirmed that Prostaglandin Endoperoxide Synthase (PTGS), also known as Cyclooxygenase-2 (COX-2), when overexpressed in OS cells, may increase resistance to tumorigenesis by elevating Reactive Oxygen Species (ROS) to a level that curtails cell viability. These key gene clusters might serve as robust predictors for OS and are crucial in the development of OS.
This study confirmed the immune modulatory effects of matrine in OS. Matrine commonly dampens immune responses of B cells and macrophages, thus modulating autoimmune reactions. Sun et al.[34] observed that, upon administering matrine to mice, there was a marked reduction in IL-6, IL-10, Tumor Necrosis Factor Alpha (TNF-α), and Interferon Gamma (IFN-γ) production within lung tissues. Concurrently, viral loads diminished significantly. Furthermore, there was an increase in the proportions of Clusters of Differentiation (CD) 4+ T cells, CD8+ T cells, and B cells in the peripheral blood compared to the control group, indicating a linkage between matrine and the regulation of immune functions. In a separate investigation, Jiang et al.[35] found that matrine treatment enhanced tight junction formation by astrocytes at glial boundaries, leading to decreased immune cell penetration into the Central Nervous System (CNS) parenchyma. Such findings suggest that the effect of matrine on OS might be channeled through macrophages, providing a potential therapeutic avenue in clinical practice.
This study has several limitations, notably the absence of in vitro assays and prospective investigations. Although the exact molecular mechanism through which matrine affects OS has not been fully elucidated[35], a major strength of our research is the detailed identification of target cells for matrine using single-cell sequencing. This insight lays a crucial foundation for future studies aiming to explore specific molecular pathways involved.
In this study, we investigated the molecular mechanisms underlying Matrine's interaction with OS, with a particular emphasis on its gene regulatory functions. Using network pharmacology, we elucidated the molecular BP associated with Matrine's therapeutic efficacy in clinical settings.
Acknowledgments:
This work was supported by the Innovation Project of Guangxi Graduate Education (No: YCBZ2023093). The Scientific and Technological Innovation Major Base of Guangxi (No: 2022-36-Z05).
Author’s contributions:
Wenkai Xie and Tianjun Ma have contributed equally to this work.
Conflict of interests:
The authors declared no conflict of interests.
References
- Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. Ca Cancer J Clin 2021;71(1):7-33.
- Yang C, Tian Y, Zhao F, Chen Z, Su P, Li Y, et al. Bone microenvironment and osteosarcoma metastasis. Int J Mol Sci 2020;21(19):6985.
[Crossref] [Google Scholar] [PubMed]
- Friebele JC, Peck J, Pan X, Abdel-Rasoul M, Mayerson JL. Osteosarcoma: A meta-analysis and review of the literature. Am J Orthop 2015;44(12):547-53.
[Google Scholar] [PubMed]
- Allison DC, Carney SC, Ahlmann ER, Hendifar A, Chawla S, Fedenko A, et al. A meta-analysis of osteosarcoma outcomes in the modern medical era. Sarcoma 2012;2012:704872.
[Crossref] [Google Scholar] [PubMed]
- Thanindratarn P, Dean DC, Nelson SD, Hornicek FJ, Duan Z. Advances in immune checkpoint inhibitors for bone sarcoma therapy. J Bone Oncol 2019;15:100221.
- Wang SD, Li HY, Li BH, Xie T, Zhu T, Sun LL, et al. The role of CTLA-4 and PD-1 in anti-tumor immune response and their potential efficacy against osteosarcoma. Int Immunopharmacol 2016;38:81-9.
[Crossref] [Google Scholar] [PubMed]
- Lussier DM, Johnson JL, Hingorani P, Blattman JN. Combination immunotherapy with α-CTLA-4 and α-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J Immunother Cancer 2015;3(1):1-11.
[Crossref] [Google Scholar] [PubMed]
- Li X, Tang Z, Wen L, Jiang C, Feng Q. Matrine: A review of its pharmacology, pharmacokinetics, toxicity, clinical application and preparation researches. J Ethnopharmacol 2021;269:113682.
[Crossref] [Google Scholar] [PubMed]
- Lin Y, He F, Wu L, Xu Y, Du Q. Matrine exerts pharmacological effects through multiple signaling pathways: A comprehensive review. Drug Des Dev Ther 2022;16:533-69.
[Crossref] [Google Scholar] [PubMed]
- Zhao B, Hui X, Wang J, Zeng H, Yan Y, Hu Q, et al. Matrine suppresses lung cancer metastasis via targeting M2-like tumour-associated-macrophages polarization. Am J Cancer Res 2021;11(9):4308.
[Google Scholar] [PubMed]
- Pu J, Tang X, Zhuang X, Hu Z, He K, Wu Y, et al. Matrine induces apoptosis via targeting CCR7 and enhances the effect of anticancer drugs in non-small cell lung cancer in vitro. Innate Immun 2018;24(7):394-9.
[Crossref] [Google Scholar] [PubMed]
- Guo S, Chen Y, Pang C, Wang X, Shi S, Zhang H, et al. Matrine is a novel inhibitor of the TMEM16A chloride channel with antilung adenocarcinoma effects. J Cell Physiol 2019;234(6):8698-708.
[Crossref] [Google Scholar] [PubMed]
- Xu GP, Zhao W, Zhuang JP, Zu JN, Wang DY, Han F, et al. Matrine inhibits the growth and induces apoptosis of osteosarcoma cells in vitro by inactivating the Akt pathway. Tumor Biol 2015;36:1653-9.
[Crossref] [Google Scholar] [PubMed]
- Zhang R, Zhu X, Bai H, Ning K. Network pharmacology databases for traditional Chinese medicine: Review and assessment. Front Pharmacol 2019;10:123.
[Crossref] [Google Scholar] [PubMed]
- Yu G, Wang LG, Han Y, He QY. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012;16(5):284-7.
[Crossref] [Google Scholar] [PubMed]
- Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling tumor infiltrating immune cells with CIBERSORT. Cancer Syst Biol 2018:243-59.
[Crossref] [Google Scholar] [PubMed]
- Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the surveillance, epidemiology, and end results program. Cancer 2009;115(7):1531-43.
[Crossref] [Google Scholar] [PubMed]
- Murphey MD, Robbin MR, MCRae GA, Flemming DJ, Temple HT, Kransdorf MJ. The many faces of osteosarcoma. Radiographics 1997;17(5):1205-31.
[Crossref] [Google Scholar] [PubMed]
- Mialou V, Philip T, Kalifa C, Perol D, Gentet JC, Marec-Berard P, et al. Metastatic osteosarcoma at diagnosis: Prognostic factors and long-term outcome-The French pediatric experience. Cancer 2005;104(5):1100-9.
[Crossref] [Google Scholar] [PubMed]
- Hu G, Cao C, Deng Z, Li J, Zhou X, Huang Z, et al. Effects of matrine in combination with cisplatin on liver cancer. Oncol Lett 2021;21(1):1-11.
[Crossref] [Google Scholar] [PubMed]
- Chen MH, Gu YY, Zhang AL, Sze DM, Mo SL, May BH. Biological effects and mechanisms of matrine and other constituents of Sophora flavescens in colorectal cancer. Pharmacol Res 2021;171:105778.
[Crossref] [Google Scholar] [PubMed]
- Wu L, Wang G, Wei J, Huang N, Zhang S, Yang F, et al. Matrine derivative YF-18 inhibits lung cancer cell proliferation and migration through down-regulating Skp2. Oncotarget 2017;8(7):11729.
[Crossref] [Google Scholar] [PubMed]
- Liang CZ, Zhang JK, Shi Z, Liu B, Shen CQ, Tao HM. Matrine induces caspase-dependent apoptosis in human osteosarcoma cells in vitro and in vivo through the upregulation of Bax and Fas/FasL and downregulation of Bcl-2. Cancer Chemother Pharmacol 2012;69:317-31.
[Crossref] [Google Scholar] [PubMed]
- Thomas DD, Lacinski RA, Lindsey BA. Single-cell RNA-seq reveals intratumoral heterogeneity in osteosarcoma patients: A review. J Bone Oncol 2023;39:100475.
[Crossref] [Google Scholar] [PubMed]
- Zhang J, Yu XH, Yan YG, Wang C, Wang WJ. PI3K/Akt signaling in osteosarcoma. Clin Chim Acta 2015;444:182-92.
[Crossref] [Google Scholar] [PubMed]
- Luo ZW, Liu PP, Wang ZX, Chen CY, Xie H. Macrophages in osteosarcoma immune microenvironment: Implications for immunotherapy. Front Oncol 2020;10:586580.
[Crossref] [Google Scholar] [PubMed]
- Lin YM, Chang ZL, Liao YY, Chou MC, Tang CH. IL-6 promotes ICAM-1 expression and cell motility in human osteosarcoma. Cancer Lett 2013;328(1):135-43.
[Crossref] [Google Scholar] [PubMed]
- Wang Q, Cai J, Wang J, Xiong C, Zhao J. miR-143 inhibits EGFR-signaling-dependent osteosarcoma invasion. Tumor Biol 2014;35:12743-8.
[Crossref] [Google Scholar] [PubMed]
- Liu Q, Wang Z, Zhou X, Tang M, Tan W, Sun T, et al. miR-485-5p/HSP90 axis blocks Akt1 phosphorylation to suppress osteosarcoma cell proliferation and migration via PI3K/AKT pathway. J Physiol Biochem 2020;76:279-90.
[Crossref] [Google Scholar] [PubMed]
- Chen CL, Zhang L, Jiao YR, Zhou Y, Ge QF, Li PC, et al. miR-134 inhibits osteosarcoma cell invasion and metastasis through targeting MMP 1 and MMP 3 in vitro and in vivo. FEBS Lett 2019;593(10):1089-101.
[Crossref] [Google Scholar] [PubMed]
- Zhang C, Zhao Y, Zeng B. Enhanced chemosensitivity by simultaneously inhibiting cell cycle progression and promoting apoptosis of drug-resistant osteosarcoma MG63/DXR cells by targeting cyclin D1 and Bcl-2. Cancer Biomarkers 2013;12(4-5):155-67.
[Crossref] [Google Scholar] [PubMed]
- Li S, Cui Z, Meng X. Knockdown of PARP-1 inhibits proliferation and ERK signals, increasing drug sensitivity in osteosarcoma U2OS cells. Oncol Res 2016;24(4):279-86.
[Crossref] [Google Scholar] [PubMed]
- Xu Z, Choudhary S, Voznesensky O, Mehrotra M, Woodard M, Hansen M, et al. Overexpression of COX-2 in human osteosarcoma cells decreases proliferation and increases apoptosis. Cancer Res 2006;66(13):6657-64.
[Crossref] [Google Scholar] [PubMed]
- Sun J, Zhao RH, Guo SS, Shi YJ, Bao L, Geng ZH et al. Effect of matrine sodium chloride injection on a mouse model combining disease with syndrome of human coronavirus pneumonia with cold-dampness pestilence attacking the lung. Acta Pharm Sin 2020:366-73.
[Crossref] [Google Scholar] [PubMed]
- Jing Y, Ma R, Chu Y, Dou M, Wang M, Li X, et al. Matrine treatment induced an A2 astrocyte phenotype and protected the blood-brain barrier in CNS autoimmunity. J Chem Neuroanatomy 2021;117:102004.
[Crossref] [Google Scholar] [PubMed]





