- *Corresponding Author:
- K. Shah
Institute of Pharmaceutical Research, GLA University, Mathura, Uttar Pradesh 281406, India
E-mail: kamal0603@gmail.com
| Date of Received | 13 November 2020 |
| Date of Revision | 18 September 2021 |
| Date of Acceptance | 20 May 2022 |
| Indian J Pharm Sci 2022;84(3):642-653 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
Cyclooxygenase inhibitors are widely used in prescription. They are the choice of drugs worldwide. The drug naproxen, which is a propionic acid nonselective cyclooxygenase inhibitor, is still preferred in arthritis, osteoarthritis and gout. The drawback associated with this drug is that it causes nausea, vomiting or gastrointestinal upsets. The cause of gastrointestinal upset may be due to perforation of gastric mucosa or ulceration. To overcome this problem, this is associated with the free carboxylic group present on naproxen. Here the parent drug is modified using glycolic acid precursors with phytophenols. The phytophenols are known for their antioxidant properties and they also reduce ulceration. The glycolic acid spacer provides a single bond rotation that promotes excellent binding with the receptor. The different derivatives of naproxen were designed and screened by computational technique based on virtual screening against human cyclooxygenase-2 enzyme through AutoDock. The identified derivatives were curtained through Lipinski’s rule of pharmacokinetics. The effective and innocuous molecule was identified. The identified derivative was synthesized, purified, characterized and pharmacological studies were done. The result obtained clearly indicating that there is a depiction in molecular docking and pharmacological studies.
Keywords
Naproxen, cyclooxygenase, gastrointestinal, glycolic acid spacer, ligands, molecular docking, inflammation, ulcer
Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) are one of the common drugs in prescription so widely used due to its analgesic and anti-inflammatory property and due to the extensive utility of NSAIDs, preferred for treatment purposes. The parent drug selected for the experiment is Naproxen (NXN). It is a non-selective Cyclooxygenase (COX) inhibitor bearing propionic acid in structure[1,2]. It is primarily given in the treatment of pain of arthritis or gout[3,4]. The drawback associated with the parent drug is the free carboxylic group which causes peripheral erosion of mucosa of the stomach which results in gastric ulcer. The non-selective nature of NXN indicates that it not only inhibits COX-2 but also COX-1. COX-1 is responsible for damaging the desired mucus lining of the stomach that results in erosion or Gastrointestinal (GI) bleeding[5,6]. Here the parent drug is modified using Glycolic acid (-OCH2COO-) precursors with phytophenols. The phytophenols are known for their antioxidant properties[7]. They act by masking through reactive oxygen species which are responsible for ulceration[8,9]. The glycolic acid spacer provides single bond rotation that promotes excellent binding with receptor. The antioxidants agents selected for the study were syringaldehyde, Menthol (MNT), sesamol, umbelliferone, thymol, carvacrol, eugenol and vanillin. The docking studies were performed with designed derivatives of NXN coupled with antioxidants through spacer against COX-2 receptor. This computational study emancipates the researchers to save resources, time and money. This study gives safe and potent derivative. The screened derivative was synthesized in the chemistry laboratory and its pharmacological studies were established.
Materials and Methods
Materials:
The three-dimensional structural model of the COX-2 enzyme complexed with the ligand mefenamic acid was downloaded from the protein data bank database and its molecular docking simulation based on in silico screening was performed by using AutoDock 4.2 software[10]. The melting point was determined using the open capillary method with the help of melting point apparatus. The Ultraviolet (UV)/ Visible spectrophotometer available in the department (Shimadzu UV-1800, quartz cells) was utilized for the experiment. The method was established for the synthesized derivative using methyl alcohol at 260 nm absorption maxima, which had a linearity range in between 5-30 μg/ml. Fourier Transform Infrared (FTIR) spectrophotometer (using DRS-8000A) of Shimadzu IRAffinity-1 made was used to record infrared spectrum. The Proton Nuclear Magnetic Resonance (1H NMR) spectrum acquired by using Bruker Avance II 400 MHz using Deuterated Chloroform (CDCl3) as the solvent. The mass spectrum of Jeol SX 102/DA-600 mass spectrometer made, using Electron Ionization (EI) technique was used to record the mass spectrum. The purity of the synthesized drug was checked by Thin Layer Chromatography (TLC) using silica gel G. Iodine vapors were used for visualization. The mobile phase used was methanol:toluene (3:7). The parent drug and MNT were obtained from Yarrow Chem Product, Mumbai 400 086, India. Analytical/spectroscopic grade chemicals were used in the whole experiment.
In silico molecular docking simulation:
The mefenamic acid was separated from the threedimensional structural complex of the human COX-2 enzyme by using Chimera software[11]. The preparation of receptor and separated ligand mefenamic acid for molecular docking was performed by using AutoDock software[10]. The grid points were enumerated by considering the ligand and all the residues interacting with it to make certain that the ligand’s extended conformations fit well in the grid box. The grid parameter file consisting of these grid points was utilized by AutoGrid utility of the AutoDock suite for generating various map files for receptor as well as ligand, which were further utilized by AutoDock for performing docking simulations[12,13].
Docking parameter file consisting of various parameters utilized for performing molecular docking of the COX-2 enzyme was prepared by AutoDock. The AutoDock software utilized the Lamarckian Genetic Algorithm (LGA) as it is the primary conformational search algorithm for performing the docking studies. The ligand’s probable binding pattern was obtained based on their position and orientations identified after the molecular docking simulations. The parameters included in the current in silico study were validated by performing docking of the human COX-2 receptor against the crystallized ligand mefenamic acid[14,15]. The docking of the COX-2 receptor was validated by considering the binding energy, chemical interaction and its overlay of docked conformation of the ligand[16,17]. The parameters used in the docking studies were validated if the binding energy of the bound ligand should be in the predefined empirical range of -5 to -15 kcal/mol[18,19], having similar binding interactions as that were present in the downloaded enzyme complex and perfect overlay of the docked conformation of the bound ligand mefenamic acid regarding its bioactive conformation[14,18].
The comparative docking study of the existing approved COX-2 inhibitors was performed to pick the parent molecule based on the best binding score as well as potential binding interactions with the residues present in the active cavity of the macromolecular target. Ibuprofen, fluprofen, fenoprofen, ketoprofen, NXN and suprofen were used in the current comparative analysis for identification of the molecule having most potent binding interaction against the COX-2 enzyme.
A ligand library consisting of 8 esteric derivatives of NXN nuclei were prepared by complexing it with some of the phytophenols like carvacrol, guaiacol, eugenol, MNT, sesamol, thymol, umbelliferone and vanillin with the intent to increase the bioavailability of the complex derivatives at the site of action[13-15]. The validated docking parameters were further utilized to perform the molecular docking simulation based on virtual screening of the designed ligands against the target COX-2 enzyme to identify its potential inhibitors. The shortlisted leads were supposed to have a high affinity for the human COX-2 and possessing potential anti-inflammatory activity. The potential leads were shortlisted by considering the minimum binding energy in the predefined empirical range and were further evaluated for their pharmacokinetics and toxic effects by using Data Warrior[20] software[12,21,22]. This tool predicted the drug-likeness and score for the leads by considering their physicochemical properties[21,22].
Synthesis:
The first step involved the reaction of a Chloroacetyl Chloride (CAC) and MNT at lower temperature. This leads to synthesize ester. The obtained ester was further reacted with NXN to get the desired derivative. The MNT (0.1 mol) with triethylamine (0.1 mol) taken in dichloromethane (50 ml) at 0°. The CAC (0.1 mol) solution made in chloroform (25 ml) was added dropwise with constant stirring for 1 h, further the reaction mixture was stirred for about 4 h. The product obtained was washed with hydrochloric acid solution (0.1 N, 3×20 ml) followed by sodium hydroxide (0.1 N, 3×20 ml) washing. The product obtained was dried and solvent was removed under pressure to get the Antioxidant Derivative (AOD). The derivative was purified by recrystallization with hot ethanol. The AOD (0.01 mol) and parent drug NXN in equimolar amount were taken in round bottom flask using solvent dichloromethane (50 ml). The reaction mixture was stirred for 24 h at Room Temperature (RT). The organic layer obtained was washed with hydrochloric acid and sodium hydroxide solutions of 0.1 N. Finally, the solvent was removed under pressure and the final product (NMXP) was recrystallized with hot ethanol (fig. 1).

Fig. 1: Synthetic scheme of NMXP
Characterization of the synthesized drug:
Solubility and partition coefficient: The solubility of the synthesized derivative was checked by using various solvents. The partition coefficient of synthesized drug was determined by using n-octanol/phosphate buffer (pH 7.4). The NMXP (1 g) was weighed and transferred to 50 ml separating funnel having equal volumes of phosphate buffer and n-octanol. The separating funnel was shaken for 2 h at RT and kept undisturbed for 1 h. The aqueous layer (10 ml) was collected and underwent extraction with dichloromethane thrice. The dichloromethane layer had undergone quantitative analysis by the UV analysis.
Kinetics study: The United States Pharmacopeia (USP) apparatus II i.e. paddle type was used for the study of kinetics. The buffer solutions i.e. phosphate buffer having pH 7.4 and hydrochloric acid buffer having pH 2 were utilized during the study at 37°. This study helped us to govern the rate of chemical hydrolysis. The synthesized derivative NMXP (1 g) was taken in 100 ml volumetric flask and made to dissolve in methanol. The NMXP solution in methanol was kept at RT for 10 min. Further NMXP solution was transferred to vessel of the dissolution apparatus comprising of 900 ml of 0.1 M hydrochloric acid. The dissolution apparatus containing drug solution was stirred at 100 rpm and a solution of 10 ml was taken out at a gap of 30 min for upto 3 h. The solution withdrawn should be replaced by fresh buffer solution instantly. The solution withdrawn should be extracted with chloroform (3×5 ml). The extracted layer was dried on desiccated sodium sulphate further chloroform was removed under pressure. The obtained residue was taken in methanol and was dried further for estimation. Similarly, kinetics studies were done in phosphate buffer (pH 7.4).
Pharmacology:
The pharmacological experiments were performed as per the recommendation and approval of the Institutional Animal Ethical Committee (IAEC) of the Institute of Pharmaceutical Research, GLA University, Mathura, Uttar Pradesh, India (1260/PO/ Re/S/09/CPCSEA). The experiments were approved by IAEC on February 23, 2019 as per the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA) guidelines. Animals (Rats and Mice) were issued from the departmental animal house. The animals were maintained at standard conditions in animal house, where the temperature was 25°±2°, fed with standard animal feed and water ad libitum, kept in 12 h dark and light cycle along with relative humidity of 45 %-55 %. The pharmacological studies including analgesic, anti-inflammatory and ulcerogenic activity of NXN derivative (NMXP) and standard drug (NXN) were done. They were evaluated for pharmacological activities based on various methods. The details are as follows:
Anti-inflammatory activity: The carrageenan induced paw edema method was utilized to judge the antiinflammatory response[23]. The experimental animals used were albino rats of wistar strains, weighing 150-200 g. The animals used for experiment were fasted overnight prior to test. The animals were divided into three groups each group comprised of six animals. The various groups were group I vehicle control, group II was of standard drug (NXN) at dose of 53 mg/kg body weight and group III was for synthesized derivative (NMXP) at the dose of 98 mg/kg body weight, 230 mM, orally (p.o.). The vehicle control animals were fed with 0.5 % Carboxymethylcellulose (CMC) solution.
Analgesic activity: The analgesic activity was determined by abdominal writhing assay method[24]. The animals used during this study were Swiss albino mice, weighing 20-25 g of either sex. Three groups were made and each comprised of six animals. The vehicle used to prepare the sample was CMC (0.5 %). The suspensions of standard drug and synthesized derivative were made. These suspensions were fed orally prior to acetic acid solution (freshly prepared, 0.6 %, 10 ml/kg). The acetic acid solution was given intraperitoneally. The writhing was counted in terms of number of constrictions of the abdomen or turning of trunk or extension of hind limbs. The readings were noted for 20 min as writhes (for each animal). The readings obtained for synthesized derivative was compared with group I (vehicle control) and group II (standard drug) animals. The degree of analgesia was expressed as percent (%) inhibition as follows:
% inhibition=(1–Wt/WC)×100
Where, Wt=Writhing in derivative/standard treated animal and WC=Writhing in group I (Vehicle) control.
Ulcerogenic study: The ulcerogenic activity was performed to confirm gastro sparing activity of synthesized derivative. The animals utilized in this study were albino wistar rats. The animals used for activity were fasted for 24 h before activity[25]. The animals were divided into three groups each group comprised of six animals. The derivative and standard drug were administered at doses of 10 times of antiinflammatory activity. These animals were sacrificed 12 h after the treatment. The ulcer protective activity was observed by taking out the stomach from the rat. Then the stomach was unbolted along the larger curvature, rinsed with sodium chloride (0.9 % w/v) solution and the reading was taken. The observations made were scored as: 0=No observable damage; 1=Superficial ulcers; 2=Deep ulcers and 3=Perforation.
Statistical analysis:
Statistical analysis was carried out for in vivo studies data. The ulcer index data was subjected to student t-test (unpaired), Analysis of Variance (ANOVA) test, followed by Dunnett’s test for determining the levels of significance in antioxidant studies, p values<0.05 were considered statistically significant.
Results and Discussion
In silico molecular docking simulation was explained here. The one out of two identical chains of 551 amino acids present in the macromolecular complex was procured by deleting another one by using Chimera software. The macromolecule was prepared for docking by removing unnecessary water molecules, the addition of polar hydrogen, followed by addition and equal distribution of Gasteiger charge. Maximum possible flexibility is provided to the ligand in the current study by keeping all the available three bonds rotatable. The crystallized structure of human COX-2 enzyme complexed with mefenamic was shown in fig. 2[18].

Fig. 2: Structure model of the crystallized human COX-2 enzyme (Green) complexed with mefenamic acid (Red)
The macromolecular residues Tyr385 and Ser530 were involved in the active interactions with mefenamic acid. The grid coordinates utilized for making threedimensional imaginary grid-box are shown in Table 1 and its docking results were tabulated in Table 2.
| Proteins | x-D | y-D | z-D | Spacing (A) | x center | y center | z center |
|---|---|---|---|---|---|---|---|
| 5IKR | 46 | 44 | 46 | 0.375 | 38.042 | 2.131 | 61.280 |
Table 1: The Grid Coordinates of the Grid-Box
| S. No. | Ligand | Structure | Binding energy (kcal/mol) | Interacting residues |
|---|---|---|---|---|
| 1 | Mefenamic acid | 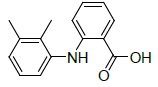 |
-7.67 | Val349, Ala527, Val116, Leu537, Ser530, Tyr385, Leu352 |
| 2 | Suprofen | 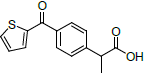 |
-6.91 | Met522, Trp387, Leu352, Val349, Leu359, Tyr355, Arg120, Val523, Ala527, Ser530 |
| 3 | Ibuprofen | 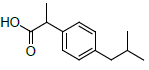 |
-6.87 | Leu352, Val349, Ala527, Val116, Ala527, Leu359, Tyr355, Arg120 |
| 4 | Fenoprofen | 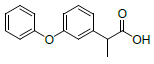 |
-6.01 | Phe205, Val344, Leu534, Leu352, Val349, Ala527, Leu531, Tyr385 |
| 5 | Fluprofen | 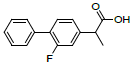 |
-7.16 | Leu352, Val349, Ala527, Tyr325, Arg120, Leu359, Val116, Leu531 |
| 6 | Ketoprofen | 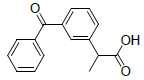 |
-5.84 | Val349, Leu531, Ala527, Tyr355, Met522, Leu352, Val523, Trp387, Ser530 |
| 7 | NXN | 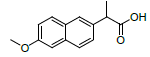 |
-7.55 | Leu359, Val523, Tyr355, Arg120, Ala527, Met522, Gly526, Leu352, Val349 |
Table 2: Comparative Study of Approved COX-2 Inhibitor Molecules
The molecular docking process of mefenamic acid against human COX-2 enzyme was successfully validated as the binding energy was -7.67 kcal/mol lies well within the predefined empirical range. The docked conformation of the ligand was having similar interactions and it was perfectly overlaid with respect to its bioactive conformation. The docked conformation having a perfect overlay with respect to its bioactive conformation was represented in fig. 3. The binding interactions of the docked ligand with reference to its bioactive crystal structure were shown in fig. 4.

Fig. 3: The superimposed docked conformation of mefenamic acid with respect to its bioactive conformation

Fig. 4: Binding mode and chemical interactions of the bound ligand mefenamic acid within the active ligand binding site of COX-2 receptor of human
The comparative analysis of the approved COX-2 inhibitors on the basis of the observed binding energy as well as their binding interactions with the macromolecular target clearly signifies NXN as the most potent COX-2 inhibitor. Thus, NXN was considered as a standard molecule in the current study and was further utilized for developing a ligand library by generating its various ester derivatives as a COX-2 inhibitor. The result of the comparative study of existing COX-2 inhibitors is shown in Table 2.
The prepared ligand library of the ester derivatives of NXN was virtually screened to identify potential COX-2 inhibitors. The structure activity relationship obtained after evaluating the binding pattern of all the lead molecules with the binding residues of the human COX-2 enzyme reveals that all the lead molecules were interacting with the Leu352, Phe518, Val349, Val523 and Ala527. Thus Leu352, Phe518, Val349, Val523 and Ala527 residues of human COX-2 enzyme were found to be key residues playing important role in the binding of the inhibitor molecule[26]. The other residues like Trp387, Tyr385, Met522, Arg120, etc. were also having a crucial role in the binding interaction of the large proportion of lead molecules. The docking results of the virtually screened ligand molecules against COX-2 enzyme were tabulated in Table 3.
| S. No. | Name | Structure | Binding energy | Interactions |
|---|---|---|---|---|
| 1 | NMXP | 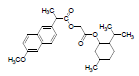 |
-8.28 | Tyr385, Leu352, Phe518, Trp387, Met522, Val523, Gly526, Ala527, Leu384, Tyr355, Leu359, Leu531, Met113, Ile345, Val116, Val349, Tyr348, Ser530, Phe381 |
| 2 | NAPE | 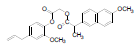 |
-7.74 | Val523, Gln192, Leu352, Met522, Phe518, Leu531, Val116, Val349, Leu359, Tyr355, Arg120, Ser353, Tyr348, Tyr385 |
| 3 | NAPC | 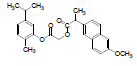 |
-7.67 | Val116, Leu359, Met113, Ile345, Leu531, Tyr355, Ser353, Ala527, Val349, Val523, Leu352, Phe518, Gly526, Met522, Trp387, Tyr385, Phe381, Phe205, Tyr348 |
| 4 | NXN | 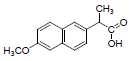 |
-7.41 | Leu359, Val523, Tyr355, Arg120, Ala527, Met522, Gly526, Leu352, Val349 |
| 5 | NAPU | 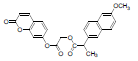 |
-7.24 | Leu531, Val349, Ala527, Leu352, Arg120, Tyr355, Val523, Ala516, Phe518, Ile517, Arg513, Ser353, Gln192, Leu359 |
| 6 | NAG | 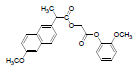 |
-7.21 | Phe518, Val523, Trp387, Leu352, Met522, Val349, Arg120, Ala527, Leu359, Leu531 |
| 7 | NAPS | 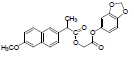 |
-6.56 | Tyr348, Phe209, Val344, Phe205, Leu534, Trp387, Tyr385, Val523, Val349, Leu352, Phe518, Leu531, Arg120, Ala527, Ser530, Phe381, Ser353, Tyr355 |
| 8 | MNT | 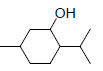 |
-5.80 | Trp387, Leu352, Val349, Ala527, Val523, Met522, Phe518, Gly526, Leu384 |
| 9 | NAPT | 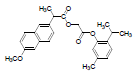 |
-5.75 | Val344, Phe205, Tyr348, Leu531Val349, Arg120, Ala527, Ser353, Leu352, Val523, Trp387, Phe381, Leu384, Tyr385, Gly526, Phe518 |
| 10 | NAPV | 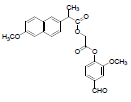 |
-5.68 | Arg120, Tyr355, Leu531, Leu359, Val116, Val349, Ala527, Leu352, Phe205, Tyr385, Ser353 |
| 11 | Thymol | 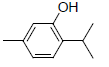 |
-5.21 | Trp387, Leu352, Leu384, Met522, Phe518, Gly526, Val523, Val349, Ala527 |
| 12 | Carvacrol | 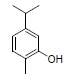 |
-5.10 | Ala527, Val349, Leu531, Leu352, Leu384, Tyr385, Val523, Trp387, Met522, Phe518, Gly526, Phe381 |
| 13 | Umbelliferone | 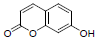 |
-5.10 | Trp382, Met522, Leu352, Tyr348, Gly526, Val523, Tyr385, Val349, Phe518 |
| 14 | Eugenol | 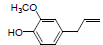 |
-4.42 | Leu384, Met522, Tyr385, Trp387, Ala527, Leu352, Val349, Phe518, Val523 |
| 15 | Vanillin | 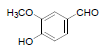 |
-4.37 | Leu352, Tyr385, Trp387, Phe381, Gly526, Ala523, Met522, Phe518 |
| 16 | Sesamol | 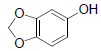 |
-4.18 | Phe357, Tyr585, Tyr547, Gln553, Trp629, His740, Ser552 |
| 17 | Guaiacol | 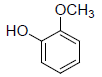 |
-3.97 | Gly526, Leu352, Phe518, Met522, Val523, Ala527 |
Table 3: Binding Energy of the Virtually Screened Leads and Their Parent Compounds against COX-2 Enzyme
All the designed NXN based ester leads were further evaluated for their pharmacokinetics by considering important physicochemical properties. The properties like calculated Partition coefficient (cLogP), Two-Dimensional Polar Surface Area (2D PSA), molecular weight, Hydrogen Bond Donor (HBD) and Hydrogen Bond Acceptor (HBA) sites etc. by using an online program OSIRIS molecular property explorer[27]. The physico-chemical properties of the shortlisted lead molecules for human COX-2 enzyme were shown in Table 4.
| Compound ID | Molecular weight | cLogP | 2D PSA (Å2) | HBA | HBD | Drug likeness |
|---|---|---|---|---|---|---|
| NXN | 230.26 | 2.99 | 46.53 | 4 | 1 | 0.361 |
| NMXP | 426 | 5.45 | 61.83 | 5 | 0 | -19.47 |
| NAPE | 434 | 5.16 | 71.06 | 6 | 0 | 1.83 |
| NAPC | 420 | 5.73 | 61.83 | 5 | 0 | -0.01 |
| NAPU | 432 | 4.04 | 88.13 | 7 | 0 | -0.78 |
| NAG | 394 | 4.13 | 71.06 | 6 | 0 | 3.5 |
| NAPS | 408 | 4.31 | 80.29 | 7 | 0 | 1.81 |
| MNT | 156 | 2.41 | 20.23 | 1 | 1 | -10.47 |
| NAPT | 420 | 5.73 | 61.83 | 5 | 0 | -0.22 |
| NAPV | 422 | 4.06 | 88.13 | 7 | 0 | 0.38 |
| Thymol | 150 | 2.84 | 20.23 | 1 | 1 | -3.02 |
| Carvacrol | 150 | 2.84 | 20.23 | 1 | 1 | -2.59 |
| Umbelliferone | 162 | 1.15 | 46.53 | 3 | 1 | -4.4 |
| Eugenol | 164 | 2.27 | 29.46 | 2 | 1 | -2.78 |
| Vanillin | 152 | 1.18 | 46.53 | 3 | 1 | -4.35 |
| Sesamol | 138 | 1.43 | 38.69 | 3 | 1 | -2.12 |
| Guaiacol | 124 | 1.24 | 29.46 | 2 | 1 | -1.68 |
Note: cLogP: Calculated partition coefficient; 2D PSA: Two-Dimensional Polar Surface Area; HBA: Hydrogen Bond Acceptor and HBD: Hydrogen Bond Donor
Table 4: “Lipinski’s Rule of Five” For the Lead Molecules Targeting COX-2 Enzyme
Later these leads were evaluated for the presence of any major toxic effect, drug-likeness and drug score values. It was observed that three out of five selected lead molecules, i.e. NMXP, N-Acetyl-D-Glucosamine (NAG) and N-Acetyl Phenylalanine derivative (NAPS) were having a good pharmacokinetic profile with the presence of no or very low toxic effects. The Absorption, Distribution, Metabolism and Excretion (ADME), and toxicity results of best lead molecules obtained after virtual screening were shown in Table 5.
| Lead compound | Mutagenic | Tumorigenic | Irritant | Reproductive effect |
|---|---|---|---|---|
| NXN | High | No | High | No |
| NMXP | No | No | No | No |
| NAPE | No | High | High | No |
| NAPC | No | No | High | No |
| NAPU | No | No | No | High |
| NAG | No | No | No | No |
| NAPS | No | No | No | No |
| MNT | High | High | High | No |
| NAPT | No | No | High | No |
| NAPV | No | No | High | No |
| Thymol | High | No | No | High |
| Carvacrol | No | No | High | No |
| Umbelliferone | High | No | No | No |
| Eugenol | High | High | High | No |
| Vanillin | High | No | High | High |
| Sesamol | No | High | No | No |
| Guaiacol | No | No | High | High |
Table 5: Toxicity Prediction of Proposed Lead Molecules for COX-2 enzyme
The derivative obtained was a resultant of nucleophilic addition/elimination reaction between acyl chlorides (acid chlorides) and alcohols followed by reaction with alkyl halide with carboxylic acid, this result in formation of the double ester separated by methylene bridge. This glycolic acid spacer resulted to orient various conformation of synthesized derivative that might allow it to have the better binding affinity at active binding site. The melting point and percentage yield of NMXP were reported as 122°-124° and 61.4 % respectively. UV (absorption maxima (λmax)) nm: Methyl alcohol-242, 260. Infrared (IR) (Potassium bromide (KBr)), cm-1: 3092.24 Aromatic C-H stretching, 2924.26 Aliphatic C-H stretching, 1716.28 C=O stretching of esters, 1114.48 C-O stretching, 1453.46 CH2- bending. 1HNMR (CDCl3, 400 MHz) Delta (δ): 1.526-1.498 (d, 3H, 3x -CH3), 1.940-1.923 (m, 1H, 4x -CH), 2.160-2.124 (d, 3H, CH3), 2.456-2.398 (m, 2H, 3x -CH2 (cyclic)), 3.458 (s, 3H, -OCH3), 3.982-3.978 (q, H, -CH-CO), 5.248 (s, 2H, -CH2 ), 7.367-7.289 (d, 6H, Ar-H). Mass Spectrometry (MS): Mass to Charge ratio (m/z) 327.320 (M+) (100 % abundance).
The oral absorption and clinical utility was manifested by calculating the value of partition coefficient and solubility data. The solubility studies were done via using various solvents (methanol, ethanol, dichloromethane, chloroform and benzene). It was found that the synthesized derivative was found to show moderate to good solubility in polar to non-polar solvents. The newly derived derivative was insoluble in water and 0.1 N hydrochloric acid, while slightly soluble in 0.1 N sodium hydroxide. This solubility parameter confirmed the hydrophobic nature of drug. The value of partition coefficient was found to be 5.46 that signify it would be a good candidate for oral absorption. This potentiates to use it orally and also designated that it had better absorption via GI tract.
The derivative synthesized was evaluated for its stability at different pH. It was known that the drug should undergo unhydrolyzed at lower pH so that it could be passed to the stomach without any hydrolysis. The synthesized drug should also show sufficient stability in intestine. The result obtained gives a clear indication that the rate of hydrolysis in acidic media was lower (4.246×10-4 s-1 and 54.48 h) than that at intestinal pH (6.436×10-3 s-1 and 39.48 h). This data completely elucidated that the synthesized derivative was stable at acidic pH.
The synthesized derivative was screened for the antiinflammatory, analgesic and ulcerogenic activities. The anti-inflammatory activity was performed by using carrageenan (1 % w/v in 1 % w/v saline solution). The carrageenan was injected in all groups of animals at the sub-plantar region of the right hind paw. The swelling of the paws was measured by using Vernier caliper. Then the readings were taken for 24 h to observe the reduction in swelling. The reduction in swelling is marked by reduction in drug treated animals in comparison with control treated animals. The data represented the synthesized derivative was found to be effective in comparison with the standard drug. The inhibition in inflammation was calculated by ratio i.e. difference of paw diameters in control and drug treated to paw diameters in control. Finally, the result obtained was compared with the standard (Table 6).
| S. No | Group | Difference in paw volume (mean±SD) | % Inhibition | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 3 h | 4 h | 6 h | 24 h | 3 h | 4 h | 6 h | 24 h | ||
| 1 | Vehicle (control) | 1.46±0.037 | 1.39±0.051 | 1.19±0.042 | 0.98±0.048 | - | - | - | - |
| 2 | NXN | 0.88±0.034* | 0.69±0.052* | 0.53±0.048* | 0.43±0.037* | 39.72 | 50.36 | 55.46 | 56.12 |
| 3 | NMXP | 0.98±0.064a | 0.84±0.210b | 0.61±0.062a | 0.48±0.043 | 34.93 | 39.57 | 48.74 | 51.02 |
Note: Data are expressed as mean±SD of six experiments; (-): Not determined; (*): p<0.0001 extremely significant as compared to control; (a): p<0.001 very significant as compared to NXN and (b): p<0.01 significant as compared to NXN
Table 6: Anti-Inflammatory Activity
The abdominal writhing method was utilized to measure the analgesic activity. The data obtained gives the idea that the synthesized derivative had retained the analgesic activity at same dose of standard drug (Table 7). The standard drug selected had ulcerogenic activity, the synthesized derivative was found to be gastric ulcer sparing. The data obtained depicted that the synthesized derivative did not cause ulceration and when it was compared with the standard drug at an equivalent dose (Table 7).
| Groups | Number of writhing | % inhibition in writhing | Ulcer index (Ui±SEM) |
|---|---|---|---|
| Control | 54.58±2.458 | - | - |
| NXN | 25.00±2.146 | 54.19±2.936 | 11.621±3.156* |
| NMXP | 28.46±3.168 | 47.85±2.241a | 7.468±4.642b |
Note: Data are expressed of six experiments; (*): p<0.0001 extremely significant as compared to control; (a): p<0.05 significant as compared to NXN; (b): p<0.01 significant as compared to NXN; Ui: Ulcer index and SEM: Standard Error of Mean
Table 7: Analgesic and Ulcerogenic Activity
Molecular docking simulation-based in silico virtual screening using Autodock was utilized in the current experimental study to design potential COX-2 targeting anti-inflammatory compounds. Three compounds NMXP, NAG and NAPS showed promising in silico results with potent inhibition of the COX-2 enzyme, good pharmacokinetic properties and the absence of any major toxic effects. These molecules could serve as promising lead compounds for further experimental validation as a novel pain-relieving drug.
The index obtained clearly signifying that the free carboxyl group present in parent drug was masked. It may be the cause of ulceration which was overcome here by this chemical modification.
In the present study NMXP drug was designed, synthesized and evaluated as a safer NSAID. The synthesized derivative was found to be chemically stable and bio labile. The synthesized drug showed desirable anti-inflammatory, analgesic activity with prominent reduced ulcerogenicity at equivalent doses. This might be due to upgraded physicochemical properties requisite for improved bioavailability. This may lead to design a safe and effective drug molecule. Based on these observations, it could be concluded that there is an advantage of giving NXN and MNT in the form of a single molecule i.e. NMXP drug.
Acknowledgements:
Authors thank to Central Instrument Facility, Bose Institute, Annex Building (First floor) Centenary campus, Kolkata for NMR and Mass data. Author also thanks the Management, GLA University, Mathura, Uttar Pradesh, India for providing the research facilities and financial assistance to carry out this work at the Institute of Pharmaceutical Research.
Conflict of interests:
The authors declare that they have no conflict of interest.
References
- Radi ZA, Khan NK. Effects of cyclooxygenase inhibition on the gastrointestinal tract. Exp Toxicol Pathol 2006;58(2):163-73.
[Crossref] [Google Scholar] [PubMed]
- Bjarnason I, Scarpignato C, Holmgren E, Olszewski M, Rainsford KD, Lanas A. Mechanisms of damage to the gastrointestinal tract from nonsteroidal anti-inflammatory drugs. Gastroenterology 2018;154(3):500-14.
[Crossref] [Google Scholar] [PubMed]
- Chan FK, Ching JY, Tse YK, Lam K, Wong GL, Ng SC, et al. Gastrointestinal safety of celecoxib versus naproxen in patients with cardiothrombotic diseases and arthritis after upper gastrointestinal bleeding (CONCERN): An industry-independent, double-blind, double-dummy, randomised trial. Lancet 2017;389(10087):2375-82.
[Crossref] [Google Scholar] [PubMed]
- Yeomans ND, Graham DY, Husni ME, Solomon DH, Stevens T, Vargo J, et al. Randomised clinical trial: Gastrointestinal events in arthritis patients treated with celecoxib, ibuprofen or naproxen in the PRECISION trial. Aliment Pharmacol Ther 2018;47(11):1453-63.
[Crossref] [Google Scholar] [PubMed]
- Kirchrath JM, Schrr K. Cyclooxygenase-2 inhibition and side-effects of non-steroidal anti-inflammatory drugs in the gastrointestinal tract. Curr Med Chem 2000;7(11):1121-9.
[Crossref] [Google Scholar] [PubMed]
- Chan N, Weitz J. In high-risk patients with arthritis and previous upper GI bleeding, celecoxib vs. naproxen reduced recurrent bleeding. Ann Intern Med 2017;167(4):17.
[Crossref] [Google Scholar] [PubMed]
- Natsume M. Polyphenols: Inflammation. Curr Pharm Des 2018;24(2):191-202.
- Sehajpal S, Prasad DN, Singh RK. Prodrugs of non-steroidal anti-inflammatory drugs (NSAIDs): A long march towards synthesis of safer NSAIDs. Mini Rev Med Chem 2018;18(14):1199-219.
[Crossref] [Google Scholar] [PubMed]
- Hassib ST, Hassan GS, El-Zaher AA, Fouad MA, Abd El-Ghafar OA, Taha EA. Synthesis and biological evaluation of new prodrugs of etodolac and tolfenamic acid with reduced ulcerogenic potential. Eur J Pharm Sci 2019;140:105101.
[Crossref] [Google Scholar] [PubMed]
- Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc 2016;11(5):905-19.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera-A visualization system for exploratory research and analysis. J Comput Chem 2004;25(13):1605-12.
[Crossref] [Google Scholar] [PubMed]
- Mujwar S, Deshmukh R, Harwansh RK, Gupta JK, Gour A. Drug repurposing approach for developing novel therapy against mupirocin-resistant Staphylococcus aureus. Assay Drug Dev Technol 2019;17(7):298-309.
[Crossref] [Google Scholar] [PubMed]
- Mujwar S, Pardasani KR. Prediction of Riboswitch as a potential drug target for infectious diseases: An in silico case study of anthrax. J Med Imaging Health Inform 2015;5(1):7-16.
- Shah K, Mujwar S, Gupta JK, Shrivastava SK, Mishra P. Molecular docking and in silico cogitation validate mefenamic acid prodrugs as human cyclooxygenase-2 inhibitor. Assay Drug Dev Technol 2019;17(6):285-91.
[Crossref] [Google Scholar] [PubMed]
- Shah K, Mujwar S, Krishna G, Gupta JK. Computational design and biological depiction of novel naproxen derivative. Assay Drug Dev Technol 2020;18(7):308-17.
[Crossref] [Google Scholar] [PubMed]
- Jain R, Mujwar S. Repurposing metocurine as main protease inhibitor to develop novel antiviral therapy for COVID-19. Struct Chem 2020;31(6):2487-99.
[Crossref] [Google Scholar] [PubMed]
- Mujwar S, Kumar V. Computational drug repurposing approach to identify potential fatty acid-binding protein-4 inhibitors to develop novel antiobesity therapy. Assay Drug Dev Technol 2020;18(7):318-27.
[Crossref] [Google Scholar] [PubMed]
- Mujwar S, Pardasani KR. Prediction of riboswitch as a potential drug target and design of its optimal inhibitors for Mycobacterium tuberculosis. Int J Comput Biol Drug Des 2015;8(4):326-47.
- Soni NU, Pardasani KR, Mujwar SO. In silico analysis of dietary agents as anticancer inhibitors of insulin like growth factor 1 receptor (IGF1R). Int J Pharm Pharm Sci 2015;7(9):191-6.
- Sander T, Freyss J, von Korff M, Rufener C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J Chem Inf Model 2015;55(2):460-73.
- Sharma KK, Singh B, Mujwar S, Bisen PS. Molecular docking based analysis to elucidate the DNA topoisomerase IIβ as the potential target for the ganoderic acid; a natural therapeutic agent in cancer therapy. Curr Comput Aided Drug Des 2020;16(2):176-89.
[Crossref] [Google Scholar] [PubMed]
- Minaz N, Razdan R, Hammock BD, Mujwar S, Goswami SK. Impact of diabetes on male sexual function in streptozotocin-induced diabetic rats: Protective role of soluble epoxide hydrolase inhibitor. Biomed Pharmacother 2019;115:108897.
[Crossref] [Google Scholar] [PubMed]
- Koster R. Acetic acid for analgesic screening. Fed Proc 1959;18:412-426.
- Winter CA, Risley EA, Nuss GW. Carrageenin-induced edema in hind paw of the rat as an assay for antiinflammatory drugs. Proc Soc Exp Biol Med 1962;111(3):544-7.
[Crossref] [Google Scholar] [PubMed]
- Shah K, Shrivastava SK, Mishra P. Synthesis, kinetics and pharmacological evaluation of mefenamic acid mutual prodrug. Acta Pol Pharm 2012;70(5):905-11.
[Google Scholar] [PubMed]
- DeLano WL. The PyMOL molecular graphics system. PyMOL by Schrodinger; 2002.
- Sander T. Molecular properties prediction-Osiris property explorer. Organic chemistry portal; 2019.





