- *Corresponding Author:
- S. K. Kashaw
Department of Pharmaceutical Sciences, Dr. Harisingh Gour University (A Central University), Sagar-470 003, India
E-mail: sushilkashaw@gmail.com
| Date of Submission | 16 December 2016 |
| Date of Revision | 05 April 2017 |
| Date of Acceptance | 09 November 2017 |
| Indian J Pharm Sci 2018;80(1):36-45 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
A New series of 1,2,4-triazole derivatives were synthesized using appropriate synthetic route and structures were confirmed by infrared spectroscopy, proton nuclear magnetic resonance, carbon-13 nuclear magnetic resonance, mass spectroscopy and elemental analysis. Antimycobacterial activity of the synthesized compounds (1-12) was carried out and percent reduction in relative light units was calculated using luciferase reporter phage assay. Percent reduction in relative light units for isoniazid was also calculated. The test compounds showed significant antitubercular potential against Mycobacterium tuberculosis H37Rv and clinical isolates, S, H, R and E resistant M. tuberculosis, when tested in vitro. Compound 8 and 12 showed better antitubercular activity compared to reference isoniazid against M. tuberculosis H37Rv strain while compounds 5, 8 and 12 found superior to isoniazid against clinical isolates: S, H, R and E resistant M. tuberculosis. Synthesized compounds were also tested in vitro against representative bacterial and fungal strains. Tested compounds showed better antibacterial activities (minimum inhibitory concentration) against Gram-positive bacteria compared to Gram-negative. Compound 5 showed better antibacterial activity than ampicillin against B. subtilis. Compound 12 in the series displayed most potent antifungal activity, which was comparable to reference fluconazole against both the fungal strains.
Keywords
1,2,4-Triazole, antibacterial, antimicrobial activity, Schiff base, isoniazid, LRP assay, Mycobacterium tuberculosis, thiol group
Although antibiotics are among the most prescribed drugs in the world today and their development and commercialization have saved countless lives, the unmet need of efficacious drugs against bacteria is still high. Several bacterial infections such as diarrhoea, food poisoning, rheumatic fever, extra intestinal and intestinal wall infections are caused by multidrug resistant bacteria [1,2]. This rapid rise in bacterial resistance towards available antibiotics is becoming a major threat to human health. Therefore, design of new class of compounds, with novel and distinct mode of action, from those of well-known classes, is of prime interest. Fungal infections pose a continuous and serious threat to human life. The severity of infection ranges from minor irritations such as athlete’s foot to life-threatening systemic infections caused by Aspergillus fumigates [3]. Hence the development of a potent, safe and selective antifungal agent is of prime importance for medicinal chemist in the quest for effective chemotherapeutic treatment for fungal diseases [4].
Tuberculosis is the leading cause of mortality from a single infectious agent and is responsible for more than three million deaths worldwide every year [5]. Despite half a century of antitubercular chemotherapy, there are still 8-10 million new cases of active tuberculosis each year and nearly two billion individuals are believed to harbour latent tuberculosis. In 2011, there were an estimated 8.7 million new cases of tuberculosis (13 % co-infected with HIV) and 1.4 million people died from tuberculosis [6]. One third of the world’s population is currently infected and more than 5000 people die from tuberculosis everyday [7]. The synergy between tuberculosis and the AIDS epidemic as well as the surge of multidrug resistant isolates of Mycobacterium tuberculosis has reaffirmed tuberculosis as a primary public health threat. The current threat in TB treatment lies in the emergence of strains resistant to two of the best antitubercular drugs, isoniazid (INH) and rifampicin (RIF).
Triazole heterocycles is a building block of great value in drug candidates [8,9] and a large number of ring systems containing this heterocyclic core have been incorporated into a wide variety of therapeutically interesting drug compounds including antiinflammatory, CNS stimulants, sedatives, antianxiety [10,11] and antimycotic agents such as fluconazole, itraconazole and voriconazole [12,13]. The 1,2,4-triazole, as well found to exhibit antiasthmatic [14], antiviral (ribavirin) [15], antifungal (fluconazole) [16], antimicrobial [17,18], antibacterial [19-21], insecticidal [22], hypnotic [23], cytotoxic [24], antitubercular [25] and hypotensive [26,27] activities. This moiety was also found in potent agonist and antagonist receptor ligands [28,29] in HIV-1 protease inhibitors [30] and in thrombin inhibitors [31].
Various isonicotinic acid derivatives like isonicotinyl hydrazides [32], isonicotinamide derivatives [33], 1,2,4-triazoles from isonicotinic acid hydrazide [34], novel nalidixic acid derived 1,2,4-triazole [35], 5-mercapto-1,2,4-triazole, novel 4H-1,2,4- triazol-3-yl-cycloalkanols [36], new imidazole and 1,2,4-triazole substituted fluorobenzimidazoles [37], and diphenylamine containing 1,2,4-triazoles [38], have been reported to possess diverse activity including antimicrobial and antitubercular. The therapeutic potential of 1,2,4-triazole prompted us to synthesize new compounds in which substituent could be arranged in a pharmacophoric pattern to display high order of antimycobacterial activity.
Driven by the findings of above documented literature, an attempt was made to incorporate various structural components to derive a novel pharmacologically active core so as to explore its potential further for antitubercular effect. The current study is such an attempt, in which 1,2,4-triazoles were synthesized and evaluated as antitubercular agents. The hypothesis was to join pyridine component of INH and Schiff base structure with the emerging antitubercular 1,2,4-triazole framework in these target compounds. The thiol group (-SH) has been introduced in the target compounds with the view that it could help binding to the cationic functional group or metal ion in the active site of the target. The compounds attempted to synthesize were given in Figure 1. Synthesized compounds were screened against sensitive and resistant M. tuberculosis strains. Additionally, these synthesized compounds were also tested in vitro for antibacterial, antifungal activities against various strains.

Figure 1: Pictorial design strategy and comparison of isoniazid and test compounds (1-12)
A: Isocratic fragment of C=O of isoniazid; B: pyridine component as in isoniazid; C: C–N–N chain resembles C–NH–NH2 of isoniazid; D: help in binding to the cationic functional group or metal ion in the active site of the target
Materials and Methods
The chemicals used for the experimental work were of synthetic grade (CDH, HiMedia, Rankem) and were used without further purification. Reactions were monitored by thin layer chromatography (TLC) on precoated silica gel-G glass plates using methanol and ethyl acetate in 3:7 ratio as mobile phase and the spots were visualized by iodine vapour. Melting points of synthesized compounds were determined by open glass capillary method and were uncorrected. Infrared (IR) spectra were recorded on Shimadzu FT-IR and Perkin-Elmer FT-IR spectrophotometer as KBr pellet and values are expressed as υmax cm-1. 1H and 13C nuclear magnetic resonance spectroscopy (NMR) spectral analysis of the synthesized compounds were recorded on a Bruker Avance III 500 MHz (AV 500) in deuterated chloroform using tetramethylsilane as internal standard. The chemical shift values were recorded on δ scale. Elemental analysis was undertaken with Elemental Vario EL III Carlo Erba 1108 analyser. Syntheses of the target compounds (1-12) were performed according to Figure 2, through following steps.

Figure 2: Synthesis of target compounds (1-12)
1 (-CH3), 2 (-CH2CH3), 3 (CH2CH2CH3), 4 (C6H5), 5 (4-F-C6H4-), 6 (4-Cl-C6H4-), 7 (4-Br-C6H4-), 8 (4-CH3-C6H4-), 9 (4-OCH3-C6H4-), 10 (-4-NO2-C6H4), 11 (-3,4,5-(OCH3)3-C6H2-), 12 (CH=CH-C6H5-)
Synthesis of potassium dithiocarbazinate (B) [39]
Isonicotinic acid hydrazide (A; 14 g, 0.10 mol) was added to a solution of potassium hydroxide (8.4 g, 0.15 mol) in absolute ethanol (100 ml) and resulting content was stirred for 10-15 min at room temperature. After this, carbon disulphide (8 ml, 0.15 mol) and ethanol (50 ml) were added to the above mixture and stirred further for 15-16 h at room temperature till the complete precipitation of potassium dithiocarbazinate (B). Precipitate thus obtained was filtered. After evaporating to dryness under reduced pressure, the residue was purified by column chromatography, eluting with methanol:ethyl acetate (30:70 v/v).
Synthesis of 4-amino-5-pyridin-4-yl-1,2,4-triazole- 3-thiol (C) [40]
Potassium dithiocarbazinate (B; 21 g, 0.079 mol), hydrazine hydrate (12 ml, 0.24 mol) and distilled water (50 ml) were placed in a round bottom flask and refluxed for 4 h, after which the reaction mixture was cooled to room temperature, diluted with cold distilled water (50 ml) and acidified with dilute acetic acid to obtain a light yellow precipitate, which, was then filtered, washed with cold distilled water, dried and recrystallized with methanol to get white crystals of 4-amino-5-pyridin-4-yl-1,2,4-triazole-3-thiol (C).
Synthesis of target compounds (1-12) [41]
A mixture of compound (C; 1g, 0.005 mol) and equimolar quantity of respective aldehydes in methanol (50-70 ml) was heated until a clear solution was obtained. This solution in presence of a few drops of concentrated hydrochloric acid was then refluxed for 3-4 h. After this reaction mixture was kept overnight at room temperature to obtain the corresponding product, which was then filtered. Evaporation and purification on silica-gel column (methanol/methylene dichloride 12:88 v/v) yielded compounds 1-12.
4-Ethylideneamino-5-pyridin-4-yl-4H-[1,2,4] triazole- 3-thiol (1)
Yield 61 % as off white solid, melting point 213-215°, IR (KBr, cm-1) υmax: 2733 (S-H), 1617 (C=N), 1121 (C-O), 702 (C-S); 1H NMR (300 MHz, CDCl3): 7.63 (m, 2H of 4-pyridine), 8.66 (m, 2H of 4-pyridine), 3.08 (s, 1H of aromatic C-SH), 7.5 (s, 1H of CH of aldimine), 1.0 (d, 3H of CH3); 13C NMR (300 MHz, CDCl3): 143.4, 122.1, 150.4, 150.4, 122.1 (C of 4-pyridine), 148.2 (C3 and C5 of triazole), 163.8 (C of N=CH), 9.33 (C of CH3); Anal. Calcd for C9H9N5S: C, 49.30; H, 4.14; N, 31.94; S, 14.62. Found: C, 49.31; H, 4.14; N, 31.96; S, 14.6. Mass (ES+) spectra of compound exhibited molecular ion peak at m/z 219 (M+).
4-Propylideneamino-5-pyridin-4-yl-4H-[1,2,4] triazole-3-thiol (2)
Yield 55 % as mud white fluffy solid, melting point 219-21°, IR (KBr, cm-1) υmax: 2725 (S-H), 1607 (C=N), 1126 (C-O), 699 (C-S); 1H NMR (300 MHz, CDCl3): 7.65 (m, 2H of 4-pyridine), 8.6 (m, 2H of 4-pyridine), 3.05 (s, 1H of aromatic C-SH), 7.53 (s, 1H of CH of aldimine), 1.4 (m, 2H of CH2), 1.1 (t, 3H of CH3); 13C NMR (300 MHz, CDCl3): 143.5, 122.5, 150.1, 150.1, 122.5 (C of 4-pyridine), 148.0 (C3 and C5 of triazole), 163.5 (C of N=CH), 18.54 (C of CH2), 10.33 (C of CH3); Anal. Calcd for C10H11N5S: C, 51.48; H, 4.75; N, 30.02; S, 13.74. Found: C, 51.47; H, 4.76; N, 30.04; S, 13.74. Mass (ES+) spectra of compound exhibited molecular ion peak at m/z 233 (M+).
4-Butylideneamino-5-pyridin-4-yl-4H-[1,2,4] triazole -3-thiol (3)
Yield 63 % as off white solid, melting point >229°, IR (KBr, cm-1) υmax: 2734 (S-H), 1611 (C=N), 1125 (C-O), 695 (C-S); 1H NMR (300 MHz, CDCl3): 7.61 (m, 2H of 4-pyridine), 8.66 (m, 2H of 4-pyridine), 3.02 (s, 1H of aromatic C-SH), 7.55 (s, 1H of CH of aldimine), 1.34 (m, 2H of CH2), 1.42 (m, 2H of CH2), 1.0 (t, 3H of CH3); 13C NMR (300 MHz, CDCl3): 143.2, 122.5, 150.2, 150.2, 122.5 (C of 4-pyridine), 148.2 and 148.1 (C3 and C5 of triazole), 163.7 (C of N=CH), 29 (C of CH2), 19.3 (C of CH2), 14.1 (C of CH3); Anal. Calcd for C11H13N5S: C, 53.42; H, 5.30; N, 28.32; S, 12.97. Found: C, 53.46; H, 5.32; N, 28.29; S, 12.92. Mass (ES+) spectra of compound exhibited molecular ion peak at m/z 247 (M+).
4-(Benzylidene-amino)-5-pyridin-4-yl-4H-[1,2,4] triazole-3-thiol (4)
Yield 58 % as brown solid, melting point 236-237°, IR (KBr, cm-1) υmax: 2720 (S-H), 1615 (C=N), 1120 (C-O), 696 (C-S); 1H NMR (300 MHz, CDCl3): 7.61 (m, 2H of 4-pyridine), 8.63 (m, 2H of 4-pyridine), 3.02 (s, 1H of aromatic C-SH), 8.09 (s, 1H of CH of N=CH), 7.60 (m, 2H of 4-aromatic CH), 7.28 (m, 3H of 4-aromatic CH); 13C NMR (300 MHz, CDCl3): 143.8, 122.5, 150.1, 150.1, 122.5 (C of 4-pyridine), 148.1 (C3and C5 of triazole), 163.8 (C of N=CH), 131.1, 129.2, 128.5, 130.3, 128.5 and 129.2 (C of 4-aryl); Anal. Calcd for C14H11N5S: C, 59.77; H, 3.94; N, 24.89; S, 11.40. Found: C, 59.75; H, 3.94; N, 24.9; S, 11.40. Mass (ES+) spectra of compound exhibited molecular ion peak at m/z 281 (M+).
4-[(4-Fluoro-benzylidene)-amino]-5-pyridin-4-yl- 4H-[1,2,4]triazole-3-thiol (5)
Yield 70 % as orange red solid, melting point 240-242°, IR (KBr, cm-1) υmax: 2737 (S-H), 1611 (C=N), 1122 (C-O), 690 (C-S); 1H NMR (300 MHz, CDCl3): 7.64 (m, 2H of 4-pyridine), 8.63 (m, 2H of 4-pyridine), 3.0 (s, 1H of aromatic C-SH), 8.08 (s, 1H of CH of N=CH), 7.62 (m, 2H of 4-aromatic CH), 7.01 (m, 2H of 4-aromatic CH); 13C NMR (300 MHz, CDCl3): 143.3, 122.9, 150.4, 150.4, 122.9 (C of 4-pyridine), 148.2 and 148.1 (C3 and C5 of triazole), 163.5 (C of N=CH), 126.7, 131.1, 116.2, 165.3, 116.2 and 131.1 (C of 4-aryl); Anal. Calcd for C14H10FN5S: C, 56.18; H, 3.37; F, 6.35; N, 23.40; S, 10.71. Found: C, 56.19; H, 3.37; F, 6.37; N, 23.40; S, 10.73. Mass (ES+) spectra of compound exhibited molecular ion peak at m/z 299 (M+).
4-[(4-Chloro-benzylidene)-amino]-5-pyridin-4-yl- 4H-[1,2,4]triazole-3-thiol (6)
Yield 78 % as orange crystalline solid, melting point 237-238°, IR (KBr, cm-1) υmax: 2731 (S-H), 1610 (C=N), 1114 (C-O), 694 (C-S); 1H NMR (300 MHz, CDCl3): 7.6 (m, 2H of 4-pyridine), 8.7 (m, 2H of 4-pyridine), 3.05 (s, 1H of aromatic C-SH), 8.05 (s, 1H of CH of N=CH), 7.63 (m, 2H of 4-aromatic CH), 7.29 (m, 2H of 4-aromatic CH); 13C NMR (300 MHz, CDCl3): 143.5, 122.5, 150.4, 150.4, 122.5 (C of 4-pyridine), 148.2 (C3 and C5 of triazole), 163.8 (C of N=CH), 130.1, 131, 130, 135.3, 130 and 131 (C of 4-aryl); Anal. Calcd for C14H10ClN5S: C, 53.25; H, 3.19; Cl, 11.23; N, 22.18; S, 10.15. Found: C, 53.22; H, 3.19; Cl, 11.25; N, 22.18; S, 10.13. Mass (ES+) spectra of compound exhibited molecular ion peak at m/z 315 (M+), 317 (M+1).
4-[(4-Bromo-benzylidene)-amino]-5-pyridin-4-yl- 4H-[1,2,4]triazole-3-thiol (7)
Yield 66 % as yellow crystalline solid, melting point 204-205°, IR (KBr, cm-1) υmax: 2730 (S-H), 1616 (C=N), 1136 (C-O), 691 (C-S); 1H NMR (300 MHz, CDCl3): 7.58 (m, 2H of 4-pyridine), 8.71 (m, 2H of 4-pyridine), 3.1 (s, 1H of aromatic C-SH), 8.0 (s, 1H of CH of N=CH), 7.53 (m, 2H of 4-aromatic CH), 7.49 (m, 2H of 4-aromatic CH); 13C NMR (300 MHz, CDCl3): 143.5, 122.3, 150.4, 150.4, 122.3 (C of 4-pyridine), 148, 148.2 (C3 and C5 of triazole), 163.6 (C of N=CH), 130.4, 131.1, 132.1, 125.3, 132.1 and 131.1 (C of 4-aryl); Anal. Calcd for C14H10BrN5S: C, 46.68; H, 2.80; Br, 22.18; N, 19.44; S, 8.90. Found: C, 46.69; H, 2.81; Br, 22.17; N, 19.44; S, 8.90. Mass (ES+) spectra of compound exhibited molecular ion peak at m/z 361 (M+), 359 (M+2).
4-[(4-Methyl-benzylidene)-amino]-5-pyridin-4-yl- 4H-[1,2,4]triazole-3-thiol (8):
Yield 75 % as yellow crystalline solid, melting point 160-162°, IR (KBr, cm-1) υmax: 2740 (S-H), 1620 (C=N), 1132 (C-O), 705 (C-S); 1H NMR (300 MHz, CDCl3): 7.62 (m, 2H of 4-pyridine), 8.65 (m, 2H of 4-pyridine), 3.15 (s, 1H of aromatic C-SH), 8.15 (s, 1H of CH of N=CH), 7.55 (m, 2H of 4-aromatic CH), 7.17 (m, 2H of 4-aromatic CH), 2.35 (s, 3H of CH3); 13C NMR (300 MHz, CDCl3): 143.3, 122.3, 150.1, 150.1, 122.3 (C of 4-pyridine), 148.2 (C3 and C5 of triazole), 163.8 (C of N=CH), 128.4, 129.3, 129.1, 140.1, 129.1 and 129.3 (C of 4-aryl); Anal. Calcd for C15H13N5S: C, 61.00; H, 4.44; N, 23.71; S, 10.86. Found: C, 61.03; H, 4.44; N, 23.71; S, 10.83. Mass (ES+) spectra of compound exhibited molecular ion peak at m/z 295 (M+).
4-[(4-Methoxy-benzylidene)-amino]-5-pyridin-4-yl- 4H-[1,2,4]triazole-3-thiol (9):
Yield 69 % as off white crystalline solid, melting point 208-210°, IR (KBr, cm-1) υmax: 2742 (S-H), 1623 (C=N), 1133 (C-O), 702 (C-S); 1H NMR (300 MHz, CDCl3): 7.61 (m, 2H of 4-pyridine), 8.67 (m, 2H of 4-pyridine), 3.08 (s, 1H of aromatic C-SH), 7.9 (s, 1H of CH of N=CH), 7.53 (m, 2H of 4-aromatic CH), 6.84 (m, 2H of 4-aromatic CH), 3.75 (s, 3H of OCH3); 13C NMR (300 MHz, CDCl3): 143.7, 122.1, 150.3, 150.3, 122.1 (C of 4-pyridine), 148, 148.1 (C3and C5 of triazole), 163.5 (C of N=CH), 123.4, 130.1, 114.2, 164.5, 114.2 and 130.1 (C of 4-aryl), 56.1 (C of OCH3); Anal. Calcd for C15H13N5OS: C, 57.86; H, 4.21; N, 22.49; O, 5.14; S, 10.30. Found: C, 57.85; H, 4.21; N, 22.47; O, 5.11; S, 10.31. Mass (ES+) spectra of compound exhibited molecular ion peak at m/z 311 (M+).
4-[(4-Nitro-benzylidene)-amino]-5-pyridin-4-yl- 4H-[1,2,4]triazole-3-thiol (10):
Yield 58 % as reddish brown powder, melting point 256-258°, IR (KBr, cm-1) υmax: 2732 (S-H), 1628 (C=N), 1136 (C-O), 692 (C-S); 1H NMR (300 MHz, CDCl3): 7.6 (m, 2H of 4-pyridine), 8.6 (m, 2H of 4-pyridine), 3.02 (s, 1H of aromatic C-SH), 8.03 (s, 1H of CH of N=CH), 7.92 (m, 2H of 4-aromatic CH), 8.31 (m, 2H of 4-aromatic CH); 13C NMR (300 MHz, CDCl3): 143.4, 122.5, 150.7, 150.7, 122.5 (C of 4-pyridine), 148.4 (C3 and C5 of triazole), 163.1 (C of N=CH), 137.1, 130, 123.5, 150.3, 123.5 and 130 (C of 4-aryl); Anal. Calcd for C14H10N6O2S: C, 51.53; H, 3.09; N, 25.75; O, 9.81; S, 9.83. Found: C, 51.55; H, 3.1; N, 25.75; O, 9.83; S, 9.83. Mass (ES+) spectra of compound exhibited molecular ion peak at m/z 326 (M+).
5-Pyridin-4-yl-4-[(3,4,5-trimethoxy-benzylidene)- amino]-4H-[1,2,4]triazole-3-thiol (11):
Yield 74 % as lemon yellow fluffy crystals, melting point 261-262°, IR (KBr, cm-1) υmax: 2737 (S-H), 1614 (C=N), 1136 (C-O), 697 (C-S); 1H NMR (300 MHz, CDCl3): 7.65 (m, 2H of 4-pyridine), 8.6 (m, 2H of 4-pyridine), 3.03 (s, 1H of aromatic C-SH), 8.0 (s, 1H of CH of N=CH), 6.53 (m, 2H of 4-aromatic CH), 3.76 (s, 9H of OCH3); 13C NMR (300 MHz, CDCl3): 143.5, 122.1, 150.5, 150.5, 122.1 (C of 4-pyridine), 148, 148.2 (C3and C5 of triazole), 163.5 (C of N=CH), 125.4, 107.1, 148.2, 134.5, 148.2 and 107.1 (C of 4-aryl), 56.5 (C of OCH3); Anal. Calcd for C17H17N5O3S: C, 54.97; H, 4.61; N, 18.86; O, 12.92; S, 8.63. Found: C, 54.99; H, 4.63; N, 18.82; O, 12.92; S, 8.63. Mass (ES+) spectra of compound exhibited molecular ion peak at m/z 371 (M+).
4-(3-Phenyl-allylideneamino)-5-pyridin-4-yl-4H- [1,2,4]triazole-3-thiol (12):
Yield 66 % as brownish yellow crystals, melting point >243°, IR (KBr, cm-1) υmax: 2733 (S-H), 1605 (C=N), 1124 (C-O), 699 (C-S); 1H NMR (300 MHz, CDCl3): 7.63 (m, 2H of 4-pyridine), 8.62 (m, 2H of 4-pyridine), 3.0 (s, 1H of aromatic C-SH), 7.54 (s, 1H of CH of N=CH), 5.56 (d, 1H of CH=CH), 6.52 (d, 1H of CH=CH), 7.32 (m, 2H of benzene), 7.23 (m, 2H of benzene), 7.15 (m, 1H of benzene); 13C NMR (300 MHz, CDCl3): 143.6, 122.3, 150.3, 150.3, 122.3 (C of 4-pyridine), 148.2, 148.1 (C3and C5 of triazole), 163.6 (C of N=CH), 112.2 and 136.1 (C of CH=CH), 134.4, 126.1, 128.7, 127.5, 128.7 and 126.1 (C of 4-aryl); Anal. Calcd for C16H13N5S: C, 62.52; H, 4.26; N, 22.78; S, 10.43. Found: C, 62.51; H, 4.24; N, 22.77; S, 10.43. Mass (ES+) spectra of compound exhibited molecular ion peak at m/z 307 (M+).
Biological evaluation
All the newly synthesized 1,2,4-triazoles were assayed in vitro for antitubercular activity against M. tuberculosis H37Rv and clinical isolates: S, H, R and E resistant M. tuberculosis. In case of antimycobacterial activity, percentage reduction in relative light units (RLU) was calculated using luciferase reporter phage (LRP) assay using INH as a reference standard. Synthesized compounds were also tested against a representative panel of bacterial and fungal pathogens by the broth microdilution minimal inhibitory concentration (MIC) method.
Fifty-microliter bacterial suspension equivalent to McFarland’s No.2 standard was added to 400 ml of G7H9 with and without the test compound. For each sample, two drug-free controls and two drug concentrations were prepared and this setup was incubated for 72 h at 37°. After incubation 50 ml of the high titer LRP (phAE129) and 400 ml of 0.1 M CaCl2 were added to all the vials and this setup was incubated at 37° for 4 h. After incubation, 100 ml of the mixture was taken from each tube into a luminometer cuvette and equal amount of working D-luciferin (0.3 mM in 0.05 M sodium citrate buffer, pH 4.5) solution was added. The RLU was measured after 10 s of integration in the luminometer (Monolight 2010). Duplicate readings were recorded for each sample and the mean was calculated. The percentage reduction in the RLU was calculated for each test sample and compared with the control. The experiment was repeated when the mean RLU of the control was less than 1000.
Antibacterial and antifungal screening
The in vitro antimicrobial activity of all the compounds and standard drugs were assessed against two representatives of Gram-positive bacteria viz. S. aureus (MTCC 96), B. subtilis (MTCC 441), two Gram-negative bacteria viz. E. coli (MTCC 1687), P. aerugenosa (MTCC 1688) and two fungi viz. C. albicans (MTCC 227), A. niger (MTCC 1344) by the broth microdilution MIC method. Mueller Hinton broth and Sabouraud dextrose broth were used as a nutrient medium to grow and dilute the compound suspension for the test bacteria and fungi, respectively. Ampicillin and norfloxacin were used as standard antibacterial drugs, whereas fluconazole was used as standard antifungal drug.
Primary inoculation of bacteria was done into Mueller- Hinton agar for overnight growth to produce a number of colonies, which were then directly suspended in saline solution until the turbidity matched the turbidity of the McFarland standard (108 CFU ml-1), i.e., inoculum size for test strain was adjusted to 108 colony forming unit (CFU)/ml per well by comparing the turbidity (turbidimetric method). Similar procedure was adopted for fungi with Sabouraud dextrose broth. Dimethyl sulfoxide (DMSO) was used as diluents to get desired concentration of the synthesized compounds and standard drugs. Each compound and standard drugs were diluted to obtain 500 μg/ml concentrations, as a stock solution. Stock solution was further progressively diluted with the test medium and required concentrations were obtained for primary and secondary screening. In primary screening 500, 250 and 125 μg/ml concentrations of the synthesized compounds were tested. The active compounds found in this primary screening were further diluted and tested against the corresponding microorganism. Each test tube was then put for incubation at 37° for 24 h for bacteria and 48 h for fungi. Growth or a lack of growth in the tubes containing the antimicrobial agent was determined by comparison with the growth control, indicated by turbidity. The lowest concentration that completely inhibited visible growth of the organism was recorded as the MIC (μg/ml). A set of tubes containing only seeded broth and the DMSO controls were maintained under identical conditions so as to make sure that the solvent had no influence on strain growth. The interpretation of the results was based on fluconazole for the fungi and also on ampicillin and norfloxacin for bacterial pathogens. The results (MIC, μg/ml) obtained are summarized in Table 2.
Results and Discussion
INH (A) was first reacted with potassium hydroxide in absolute ethanol by simple stirring the mixture at room temperature. Carbon disulphide and ethanol were then added to the above mixture and resulting content was stirred further for 15-16 h at room temperature to obtain potassium dithiocarbazinate (B). After this a mixture of potassium dithiocarbazinate (B), hydrazine hydrate and distilled water was refluxed for approximately 4 h and then cooled to room temperature, diluted with cold distilled water and acidified with dilute acetic acid to obtain a light-yellow precipitate, which appeared as white crystals of 4-amino-5-pyridin-4-yl-1,2,4- triazole-3-thiol (C) after filtration, washing with cold water, drying and recrystallization. Finally, a mix of compound (C) and equimolar quantity of respective aldehyde in methanol was then refluxed for 3-4 h in presence of concentrated hydrochloric acid and kept overnight to obtain the corresponding products (1-12).
Structures of compounds 1-12 were confirmed by IR, NMR spectroscopy data as well as their distinct Rf values in TLC analysis. As the desired target compounds (1-12) are formed, strong band of -NH2 stretch (single band in the range 3350-3500 cm-1) is disappeared and –C=N signal appear in the ranges 1605-1630 cm-1 in their corresponding IR spectra. Similarly characteristic proton NMR signal near 7.5 for 1H of CH of aldimine further confirm the formation of the target compounds. Number of signals in 13C NMR spectra of compounds 1-12 was measured with their corresponding chemical shift values to characterize the formation of compounds. Compound 1 provided nine 13C NMR signals with characteristics 163.8 for C of N=CH and 9.33 for C of CH3 attached to aldimine carbon. Similarly, compound 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12 given 10, 11, 14, 14, 14, 14, 15, 15, 14, 17 and 16 13C NMR signals along with characteristic range of aldimine C signal, which is given with individual compounds in experimental section.
All the newly synthesized compounds were assayed in vitro for antitubercular activity against M. tuberculosis H37Rv and clinical isolates: S, H, R and E resistant M. tuberculosis. In case of antimycobacterial activity, percent reduction in RLU was calculated using LRP assay at two different concentrations (50 and 100 μg/ml) using INH as a reference standard and the observed percent inhibition has been tabulated in Table 1. Compound is considered to be an antimycobacterial, if 50 % reduction in the RLU was observed when compared to the control using a luminometer.
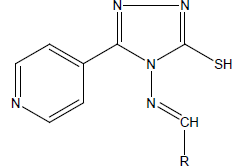 |
|||||
| Compound | R | Percent reduction in RLU | |||
| M. tuberculosisH37Rv | *Clinical isolates, S, H, R and E resistant M. tuberculosis | ||||
| 50 µg/ml | 100 µg/ml | 50 µg/ml | 100 µg/ml | ||
| 1 | -CH3 | 32.13 | 39.76 | 26.43 | 31.3 |
| 2 | -CH2CH3 | 35.3 | 43.28 | 25.0 | 32.53 |
| 3 | -CH2CH2CH3 | 39.53 | 48.71 | 29.86 | 35.51 |
| 4 | -C6H5 | 56.11 | 68.36 | 39.64 | 44.32 |
| 5 | 4-F-C6H4- | 61.14 | 80.50 | 45.97 | 50.72 |
| 6 | 4-Cl-C6H4- | 52.16 | 77.33 | 35.55 | 39.85 |
| 7 | 4-Br-C6H4- | 20.93 | 35.90 | 11.02 | 36.64 |
| 8 | 4-CH3-C6H4- | 80.54 | 82.01 | 37.36 | 52.79 |
| 9 | 4-OCH3-C6H4- | 9.72 | 25.73 | 22.79 | 41.20 |
| 10 | 4-NO2-C6H4- | 51.77 | 67.65 | 27.40 | 37.6 |
| 11 | 3,4,5-(OCH3)3- C6H2- | 10.70 | 46.08 | 6.25 | 22.74 |
| 12 | CH=CH-C6H5 | 74.07 | 83.49 | 38.07 | 52.47 |
| Isoniazid | - | 81.91 | 34.44 | ||
Table 1: Percent reduction in relative light units (RLU) against M. Tuberculosis h37RV and clinical isolates, S, H, R, and E resistant M. Tuberculosis
At 50 μg/ml concentration, compounds 4, 5, 6, 8, 10 and 12 were found to be active, while at higher concentration (100 μg/ml), these compounds showed higher activities against M. tuberculosis H37Rv. Among these, compound 8 and 12 at 100 μg/ml exhibited greater reduction in RLU than the reference standard INH, while compound 5 at 100 μg/ml displayed reduction in RLU comparable to that of the INH. Most active compounds 8, 12 and 15 of this series bear 4-CH3-C6H4-, CH=CH-C6H5- and 4-F-C6H4- substituents, respectively at 4th position in the triazole ring.
In case of clinical isolates: S, H, R, and E resistant M. tuberculosis, <50 % reduction in RLU was only seen with compounds 8, 12 and 5 at 100 μg/ml concentration, however, compounds 4, 5, 8, 12 at 50 μg/ml and 3, 4, 6, 7, 9 at 100 μg/ml showed higher reduction in RLU than INH and hence exhibited better antitubercular activity than the standard drug. Three compounds, 8, 12 and 5 found to be most potent among the series against H37Rv as well as S, H, R and E resistant M. tuberculosis strains. Higher potencies of these compounds suggest that substituents at 4th position contributing pharmacokinetically toward antitubercular activity. This has been stated because compounds 12 and 8 seems to be highly lipophilic due to contribution by styryl and tolyl components respectively, while compound 5 bears 4-F-C6H4- at 4th position that is comparatively polar in nature.
All the newly synthesized compounds were assayed in vitro for antibacterial and antifungal activity against a representative panel of bacterial and fungal pathogens. In primary screening 500, 250 and 125 μg/ml concentrations of the synthesized compounds were tested. The active compounds found in this primary screening were further diluted and tested against the corresponding microorganism to obtain their MICs, where the lowest concentration that completely inhibited visible growth of the organism was recorded as MIC (μg/ml).
The obtained results (Table 2) revealed that all of the synthesized compounds exhibited potent antibacterial activities against both Gram-positive strains. In case of Staphylococcus aureus, compound 5 (MIC=12.5 μM), 6 (MIC=14.1 μM), 9 (MIC=18.75 μM), 10 (MIC= 13.3 μM) and 11 (MIC=15.6 μM) exhibited MICs as good as ampicillin (MIC=10.2 μM). Interestingly, the same compounds i.e. 5, 6, 9, 10 and 11 were shown to be most active against Bacillus subtilis, where compound 5 gave better anti B. subtilis activity (MIC=10.2 μM) than ampicillin (MIC=10.9 μM). Antibacterial activities of the compounds improve as the electron withdrawing or donating capacity of the substituent on phenyl ring at fourth position of 1,2,4-triazole increase.
| Compound | R | Minimum inhibitory concentration (MIC, μg/ml) | |||||
|---|---|---|---|---|---|---|---|
| Gram-positive bacteria | Gram-negative bacteria | Fungi | |||||
| S. aureus | B. subtilis | P. aeruginosa | E. coli | A. niger | C. albicans | ||
| 1 | -CH3 | 62.5 | 68.8 | 34.4 | 40.6 | 28.1 | 18.8 |
| 2 | -CH2CH3 | 75 | 62.5 | 62.5 | 68.8 | 162.5 | 150 |
| 3 | -CH2CH2CH3 | 93.8 | 87.5 | 21.9 | 25 | 112.5 | 137.5 |
| 4 | -C6H5 | 31.3 | 28.1 | 14.1 | 15.6 | 17.2 | 15.6 |
| 5 | 4-F-C6H4- | 12.5 | 10.2 | 106.3 | 100 | 125 | 106.3 |
| 6 | 4-Cl-C6H4- | 14.1 | 15.6 | 87.5 | 106.3 | 118.8 | 112.5 |
| 7 | 4-Br-C6H4- | 25 | 23.4 | 75 | 62.5 | 150 | 125 |
| 8 | 4-CH3-C6H4- | 46.9 | 50 | 37.5 | 31.3 | 15.6 | 14.1 |
| 9 | 4-OCH3-C6H4- | 18.75 | 17.2 | 50 | 56.3 | 40.6 | 50 |
| 10 | 4-NO2-C6H4- | 13.3 | 12.5 | 125 | 137.5 | 100 | 87.5 |
| 11 | 3,4,5-(OCH3)3- C6H2- | 15.6 | 20.3 | 81.3 | 75 | 34.4 | 43.8 |
| 12 | -CH=CH-C6H5 | 75 | 81.3 | 93.8 | 100 | 11.7 | 10.9 |
| Ampicillin | - | 10.2 | 10.9 | - | - | - | - |
| Norfloxacin | - | 11.7 | 14.1 | 10.2 | 7.8 | - | - |
| Fluconazole | - | - | - | - | - | 9.4 | 10.2 |
Table 2: In Vitro Antibacterial and Antifungal Activity Results of Compounds 1-12
Prepared compounds showed lower antibacterial potencies against Gram-negative bacteria as compared to Gram-positive bacterial strains. Compounds displayed MICs range from 14.1 to 106.3 μM against Pseudomonas aeruginosa and from 15.6 to 137.5 μM against Escherichia coli. This may be due to the polar nature of the substituents on phenyl ring at fourth position of 1,2,4-triazole which finally turn into the lesser lipophilic compounds.
The obtained results for antifungal activities as depicted in Table 2 revealed that most of compounds could effectively inhibit the growth of the tested fungal strains, however, none of them shown to be superior to the reference drug fluconazole. Compound 12 displayed most potent antifungal activities against both fungus. Compared to the reference fluconazole (A. niger: MIC=9.4 μM; Candida albicans: MIC= 10.2 μM), compound 12 exhibited comparable (A. niger: MIC=11.7 μM; C. albicans: MIC=10.9 μM) activities. Apart from this compound 8 (A. niger: MIC=15.6 μM; C. albicans: MIC=14.1 μM) and compound 1 (A. niger: MIC=28.1 μM; C. albicans: MIC=18.8 μM) were also found to be potent antifungal.
Many significant facts were observed on analysing the effect of the substituents on the antibacterial activity of the synthesized compounds. Antibacterial activity of the compounds has been improved by the substitution of the more electron withdrawing group on the phenyl ring at the fourth position of 1,2,4-triazoles. The polar nature of the substituent on the phenyl ring at the fourth position converted the compounds into less lipophilic character. The addition of the benzyl substitution on the 1,2,4-triazoles potentiates antibacterial activity. Substitution of –F on the phenyl group potentiate the activity as compared to the ampicillin. Table 2 represent all the data of antibacterial and antifungal activity correlated with this conclusion. Substitution of methyl, ethyl, propyl and phenyl at the fourth position diminished the antibacterial activity. As far as antitubercular activity is concern substitution of more electron withdrawing group at the fourth position on the phenyl ring of the 1,2,4-triazoles is favourable for the activity. Compound 5 with 4-fluorophenyl group at position 4 of 1,2,4-triazoles exhibited better activity than phenyl or chlorophenyl substitutions. This is due to more electronegativity of fluorine than chloro. Substitution of methyl on 4th position of phenyl ring as in compound 8 has shown better activity profile as compared to electron donating group with less steric hindrance effect. The group contributes to positive inductive effect to the benzyl electron cloud leading more active electron cloud system, which has led to a positive denotable effect. Compounds with methoxy (compound 9), tri-methoxy (compound 11) or nitro (compound 10) group on phenyl ring exhibited lesser activity may be due to high steric and moderate electron withdrawing effect. Higher potencies of these compounds suggest that substituents at 4th position also contributes pharmacokinetically toward antitubercular activity because compounds 12 and 8 seems to be highly lipophilic due to contribution of styryl and tolyl components, respectively, while compound 5 bears 4-fluorophenyl at 4th position is comparatively polar in nature.
In the present investigation, 12 different 1,2,4-triazoles were synthesized and evaluated for their antimycobacterial activity. Most compounds exhibited significant antimycobacterial activity. A remarkable activity against M. tuberculosis H37Rv was found in compound 8, 12 and 5 carrying 4-CH3- C6H4-, CH=CH-C6H5- and 4-F-C6H4- substituent, respectively. Compounds 8, 12 and 5 showed more than 50 % reduction in RLU against clinical isolates, S, H, R and E resistant M. tuberculosis at 100 μg/ml concentration, however, compounds 4, 5, 8, 12 at 50 μg/ml and 3, 4, 6, 7, 9 at 100 μg/ml exhibited more reduction in RLU than INH. Overall, compounds 8, 12 and 5 found to be most potent among the series against H37Rv as well as S, H, R and E resistant M. tuberculosis strains. Synthesized compounds were found to be more active against Gram-positive bacteria as compared to Gram-negative bacteria, which might be due to highly polar substitutions in most of the compounds. Antibacterial activities against Gram-positive bacteria were found to improve with both electron withdrawing and donating substituent. Compound 5 with 4-F-C6H4- substitution showed better antibacterial activity (MIC= 10.2 μM) than ampicillin (MIC=10.9 μM) against B. subtilis. Compound 12 in the series displayed most potent antifungal activities (comparable to reference fluconazole) against both fungal strains.
Acknowledgements
One of the authors Richa Singh is grateful to University Grant Commission, New Delhi for Junior Research Fellowship.
Conflict of interest
The authors have declared no conflict of interest.
Financial support and sponsorship
Nil.
References
- Gold HS, Moellering RC Jr. Antimicrobial-drug resistance. N Engl J Med 1996;335:1445-53.
- Davies J. Inactivation of antibiotics and the dissemination of resistance genes. Science 1994;264:375-82.
- Uchida T, Somada A, Kagoshima Y, Oida S. Carbon analogues of antifungal dioxane-triazole derivatives: Synthesis and in vitroactivities. Bioorg Med ChemLett 2008;18:6538-41.
- Rezaei Z, Khabnadideh S, Pakshir K, Hossaini Z Amiri F, Assadpour E. Design, synthesis and antifungal activity of triazole and benzotriazole derivatives. Eur J Med Chem 2009;44:3064-67.
- Snider DE, Raviglione M, Kochi A, Bloom B. Tuberculosis: Pathogenesis, Protection and Control: Global Burden of Tuberculosis. Washington DC: ASM Press; 1994.
- http://apps.who.int/iris/bitstream/10665/259366/1/9789241565516-eng.pdf.
- http://www.who.int/tb/publications/global_report/en/.
- Ma LY, Pang LP, Wang B, Zhang M, Hu B, Xue DQ, et al. Design and synthesis of novel 1,2,3-triazole-pyrimidine hybrids as potential anticancer agents. Eur J Med Chem 2014;86:368-80.
- Mavrova AT, Wesselinova D, Tsenov JA, Lubenov LA. Synthesis and antiproliferative activity of some new thieno[2,3-d] pyrimidin-4(3H)-ones containing 1,2,4-triazole and 1,3,4-thiadiazole moiety. Eur J Med Chem 2014;86:676-83.
- Nadkarni BA, Kamat VR, Khadse BG. Synthesis and anthelmintic activity of 3,6-disubstituted-7H-s-triazolo(3,4-b)(1,3,4)thiadiazines. ArzneimittelForschung 2001;51:569-73.
- Isloor AM, Kalluraya B, Shetty P. Regioselective reaction: synthesis, characterization and pharmacological studies of some new Mannich bases derived from 1,2,4-triazoles. Eur J Med Chem 2009;44:3784-87.
- Budavari S. The Merck index: an encyclopedia of chemicals, drugs, and biologicals 12th ed. Whitehouse Station, New Jersey: Merck and Co. Inc; 1996.
- Haber J. Present status and perspectives on antimycotics with systemic effects. CasLekCesk 2001;140:596-604.
- Naito Y, Akahoshi F, Takeda S, Okada T, Kajii M, Nishimura H, et al. Synthesis and pharmacological activity of triazole derivatives inhibiting eosinophilia. J Med Chem 1996;39:3019-29.
- De Clercq E. Antiviral drugs in current clinical use. J ClinVirol 2004;30:115-33.
- Collin X, Sauleau A, Coulon J. 1,2,4-Triazolo mercapto and aminonitriles as potent antifungal agents. Bioorg Med ChemLett 2003;13:2601-05.
- Kidwai M, Sapra P, Misra P, Saxena RK, Singh M. Microwave assisted solid support synthesis of novel 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazepines as potent antimicrobial agents. Bioorg Med Chem 2001;9:217-20.
- Suresh Kumar GV, Rajendra Prasad Y. Chandrashekar SM. Synthesis and pharmacological evaluation of novel 4-isopropylthiazole-4-phenyl-1,2,4-triazole derivatives as potential antimicrobial and antitubercular agents. Med Chem Res 2013;22:938-48.
- Papakonstantinou-Garoufalias S, Pouli N, Marakos P, Chytyroglou-Ladas A. Synthesis antimicrobial and antifungal activity of some new 3-substituted derivatives of 4-(2,4-dichlorophenyl)-5-adamantyl)-1H-1,2,4-triazole. Farmaco 2002;57:973-77.
- Ledeti I, Bercean V, Alexa A, Foica C, Futa LM, Dehelean C, et al. Preparation and Antibacterial Properties ofSubstituted 1,2,4-Triazoles.J Chem 2015;2015:879343.
- Joshi SD, Dixit SR, More UA, Rai S, Kulkarni VH. Molecular modeling, synthesis, antibacterial and antitubercularactivities of some novel pyrrolyl 1,2,4-triazole derivatives. Indo Am J Pharm Res 2014;4(05):2323-38.
- Ghorab MM, Abdel-Hamide SG, El-Gaby MSA, El-Sayed SM. Synthesis and effect of some new [1,2,4]-Triazolo-[4,3-a]quinazolin-5(4H)-ones and related compounds on Ehrlich Ascites carcinoma cells. Acta Pharm 1999;49:1-10.
- Hester JB, Rudzik AD, Kamdar BV. 6-phenyl-4H-s-triazolo[4,3-a][1,4]benzodiazepines which have central nervous system depressant activity. J Med Chem 1971;14:1078-81.
- Milton NGN. Inhibition of catalase activity with 3-amino-triazole enhances the cytotoxicity of the Alzheimer’s amyloid-β peptide. Neurotoxicol 2001;22:767-74.
- Desai NHP, Bairwa R, Kakwani M, Tawari N, Ray MK, Rajan MG, et al. Novel 4H-1,2,4-triazol-3-yl cycloalkanols as potent antitubercular agents. Med Chem Res 2013;22:401:8.
- Burell G, Evans JM, Hadley MS, Hicks F, Stemp G. Benzopyran potassium channel activators related to cromakalim - heterocyclic amide replacements at position 4. Bioorg Med ChemLett 1994;4:1285-90.
- Ghorab MM, Abdel Hamide SG, Ali GM, El-Sayed H. Synthesis and Insecticidal Activity of Some New 3[4(3H)-Quinazolinone-2-yl)thiomethyl]-1,2,4-triazole-5-thiols. J PesticSci 1996;48:31-5.
- Wadsworth JH, Jenkins SM, Orlek BS, Cassidy F, Clark MSG, Brown F, et al. Synthesis and muscarinic activities of quinuclidin-3-yltriazole and tetrazole derivatives. J Med Chem 1992;35:1280-90.
- Chen C, Dagnino R, Huang CQ, McCarthy JR, Grigoriadis DE. 1-Alkyl-3-amino-5-aryl-1H-[1,2,4]triazoles: novel synthesis via cyclization of N-Acyl-S-methylisothioureas with alkylhydrazines and their potent corticotropin-Releasing factor-1 (CRF1) receptor antagonist activities. Bioorg Med ChemLett 2001;11:3165-68.
- Thompson SK, Eppley AM, Frazee JS, Darcy MG, Lum RT, Tomaszeck TA, et al. Synthesis and antiviral activity of a novel class of HIV-1 protease inhibitors containing a heterocyclic P1′-P2′ amide bond isostere. Bioorg Med ChemLett 1994;4:2441-46.
- Hearn MJ, Cynamon MH, Chen MF, Coppins R, Davis J, et al. Preparation and antitubercular activities in vitroand in vivo of novel Schiff bases of Isoniazid. Eur J Med Chem 2009;44:4169-78.
- Sriram D, Yogeeswari P, Priya DY. Antimycobacterial activity of novel N-(substituted)-2-isonicotinoylhydrazinocarbothioamide endowed with high activity towards isoniazid resistant tuberculosis. Biomed Pharmcother 2009;63:36-39.
- Bayrak H, Demirbas A, Demirbas N, Sengul AK. Synthesis of some new 1,2,4-triazoles starting from isonicotinic acid hydrazide and evaluation of their antimicrobial activities. Eur J Med Chem2009;44:4362-66.
- Aggarwal N, Kumar R, Dureja P, Khurana JM. Synthesis, antimicrobial evaluation and QSAR analysis of novel nalidixic acid based 1,2,4-triazole derivatives. Eur J Med Chem2011;46:4089.
- Pattan S, Gadhave P, Tambe V, Dengale S, Thakur D, Hiremath SV, et al. Synthesis and evaluation of some novel 1,2,4-triazole derivatives for antimicrobial, anti-tubercular, anti-inflammatory activities. Indian J Chem 2012;51B:297-301.
- Nandha B, Nargund LVG, Nargund SL. Design and synthesis of some new imidazole and 1,2,4-triazole substituted fluorobenzimidazoles for antitubercular and antifungal activity. Der Pharma Chemica 2013;5(6):317-27.
- Mohan Krishna K, Inturi B, Pujar GV, Purohit MN, Vijaykumar GS. Design, synthesis and 3D-QSAR studies of new diphenylamine containing 1,2,4-triazoles as potential antitubercular agents. Eur J Med Chem 2014;84:516-29.
- Fan Z, Yang Z, Zhang H, Mi N, Wang H, Cai F,et al.Synthesis, Crystal Structure, and Biological Activity of 4-Methyl-1,2,3-thiadiazole-Containing 1,2,4-Triazolo[3,4-b][1,3,4] thiadiazoles. J Agric Food Chem 2010;58(5):2630-36.
- Joshi SD, Vagdevi HM, Vaidya VP, Gadaginamath GS. Synthesis of new 4-pyrrol-1-yl benzoic acid hydrazideanalogs and some derived oxadiazole, triazole and pyrrole ring systems: A novel class of potential antibacterial and antitubercular agents. Eur J Med Chem 2008;43:1989-96.
- Bayrak H, Demirbas A, Demirbas N, Karaoglu SA. Synthesis of some new 1,2,4-triazoles starting from isonicotinic acid hydrazide and evaluation of their antimicrobial activities. Eur J Med Chem 2009,44:4362-66.
- Duncia JV, Santella JB, Higley CA, VanAtten MK, Weber PC, Alexander RS, et al. Pyrazoles, 1,2,4-triazoles, and tetrazoles as surrogates for cis-amide bonds in boronate ester thrombin inhibitors. Bioorg Med ChemLett 1998;8:775.
- Hatfull GF, Jacobs JR Jr. Mycobacteriophages: cornerstones of mycobacterial research In: Bloom B, editor. Tuberculosis: Pathogenesis, Protection and Control. Washington DC: ASM Press; 1994. p.165-83.
- Sivakumar PM, Seenivasan SP, Kumar V, Doble M. Synthesis, antimycobacterial activity evaluation, and QSAR studies of chalcone derivatives. Bioorg Med ChemLett 2007;17:1695-700.





