- *Corresponding Author:
- L. Kumar
Department of Pharmaceutics, National Institute of Pharmaceutical Education and Research, Vaishali, Bihar 844 102, India
E-mail: lk.kundlas@gmail.com
| Date of Received | 12 September 2021 |
| Date of Revision | 08 February 2023 |
| Date of Acceptance | 18 May 2023 |
| Indian J Pharm Sci 2023;85(3):678-685 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
Hesperetin is a flavanone glycoside abundantly available in citrus fruits. Hesperetin is known to have various pharmacological activities, including anti-oxidant, anti-inflammatory, anti-allergic etc. In this current study, a simple, isocratic, fast, accurate and robust high-performance liquid chromatographic analytical method was developed for the estimation of hesperetin in a topical gel. A good chromatographic separation was achieved with an inertsil ODS 3V column with acetonitrile and acidified water as mobile phase. The method was validated over a linearity range of 25 to 2500 ng/ml following regulatory guidelines for various parameters, including specificity, system suitability, linearity, accuracy, precision and robustness. A topical gel containing hesperetin was prepared using Carbopol as a gel-forming agent. The total drug content of this formulation was determined using the validated high-performance liquid chromatographic method. The validated method was found to be linear over the selected range. Limit of detection and limit of quantitation was calculated as 1.61 and 4.89 ng/ml respectively. The method was found to be precise with % relative standard deviation of less than 1 % for intraday precision and less than 2 % for inter-day precision experiments. The method was also found to be robust, with small and deliberate changes in various method parameters. The drug content of topical gel formulation was found to be more than 98 % when analyzed using the validated high-performance liquid chromatographic method. The developed method will be useful for the routine quantitative analysis of hesperetin in topical gel formulations.
Keywords
Hesperetin, chromatography, topical formulation, method validation
Flavonoids are a large group of phenolic compounds that are widely spread in plants[1]. Hesperetin is a predominant flavanone that belongs to the group of flavonoids[2]. Hesperetin is chemically known as 5,7,30-trihydroxy-40-methoxy flavanone[3]. The molecular weight of hesperetin is 302.28 g/mol with molecular formula C16H14O6. Physically hesperetin is solid with light yellow to light tan yellow powder with a faint fatty vanillic aroma. Hesperetin is soluble in ethanol, sparingly soluble in water, and log P is 2.60[4]. Hesperedin is the glycoside form of hesperetin that is easily available in citrus fruits and acts as the prodrug to hesperetin. The deglycosylation of hesperidin by intestinal bacteria results in the formation of hesperetin before its absorption[5]. Hesperetin is known to have various pharmacological activities such as anti-inflammatory, anti-oxidant, anti-microbial, anti-allergic, and anticancer activities[2,6]. From recent studies, it is evident that hesperetin has the potential to suppress tumor progression of lung adenocarcinoma in combination with platinum[2]. Hesperetin is effective in improving renal functions of diabetic rats by decreasing serum creatinine, uric acid levels, and urinary protein excretion and is effective in protecting the kidney from cisplatin-induced kidney damage[1,7]. Recently, Jayaraman et al.[3], reported antihyperglycemic and antihyperlipidemic potentials of hesperetin in a chemically induced diabetic rat model. Hesperetin has potential to combat the molecular alterations and toxicities induced by toxic heavy metals[8]. Further, literature supports that hesperetin is a potential candidate in the treatment of neurodegenerative diseases[9].
Several analytical and bioanalytical methods were developed by various researchers for the quantification of hesperetin in various types of matrices. Kanaze et al.[10] developed the High- Performance Liquid Chromatography (HPLC) method for the quantification of hesperetin along with other components in human urine samples with retention a time of more than 16 min. Recently, Saifullah et al.[11] used an HPLC analytical method for the quantification of gallic acid and hesperetin. In this study, gradient method was used for the separation of the components from lemon myrtle extract[11]. Recently, Wang et al.[12] reported a gradient method for separating hesperetin along with five other components in a citrus fruit extract. In this current study, a simple, isocratic, fast, sensitive and robust method was developed for the estimation of hesperetin in analytical samples. Further, this method was validated following regulatory guidelines. A topical gel containing hesperetin was developed using carbopol as a gelling agent. For the determination of drug content in this formulation, hesperetin was extracted from this gel and analyzed using the validated method.
Materials and Methods
Chemicals and reagents:
Hesperetin was purchased from Sigma-Aldrich, Bangalore, India. HPLC grade methanol and acetonitrile, orthophosphoric acid (OPA) (88 %), carbopol, methyl, and propyl paraben were purchased from SD Fine Chem Pvt. Ltd, India. Type-I water was obtained from Millipore Direct-Q® 3 water purification system, Millipore Corporation, MA, USA.
Instrumentation:
HPLC system (LC 2010 CHT), Shimadzu, Japan, equipped with a degasser, quaternary pump, column oven, autosampler and Ultraviolet (UV) detector, was used for the chromatographic analysis. Data acquisition, chromatographic peak integration, and surveillance were controlled by lab solutions (version 5.7) software.
Chromatographic method development:
Different mobile phase combinations and columns were screened for the separation of hesperetin, considering chromatographic parameters such as peak area, Tailing Factor (TF), Number of Theoretical Plates (NTP), and capacity factor (k). A combination of acetonitrile and acidified water (pH adjusted to 3.0 with OPA) was used as the mobile phase. Intersil ODS 3V column (250 mm length, 4.6 mm internal diameter, 5 μ particle size, 100 Å pore size) was used as the stationary phase. Mobile phase [acetonitrile:OPA in water, pH, 3.0 (50:50, % v/v)] was pumped at a flow rate of 1.200 ml/min on stationary phase adjusting column oven temperature to 25° and autosampler to 4°. The effluent was monitored with UV detector and wavelength fixed at 288 nm.
Preparation of stock and working solutions:
An accurate amount of hesperetin was weighed and dissolved in methanol to achieve a concentration of 1 mg/ml. From this stock, working solutions consisting of 8 standards were prepared serially using methanol over a range of 25 to 2500 ng/ml. All the working solutions and stock solutions were stored at 2- 8° till further use.
Method validation:
The HPLC analytical method developed for the quantification of hesperetin was validated for various parameters that include specificity, system suitability, linearity, precision, accuracy, Limit Of Detection (LOD) and Limit Of Quantitation (LOQ) and robustness. Guidelines of the United States Food and Drug Administration (US FDA) and International Conference on Harmonisation Q2R (1) (ICH) were followed for method validation[13,14].
Specificity:
Specificity is the ability of the analytical method to estimate the analyte in the presence of other components that are expected to be present in the sample. A blank sample was injected into HPLC with the developed method to check the presence of any interference at the retention time of the analyte. The peak area of the interference was recorded, and the percent interference was calculated using the formula.
Percentage of interference=Pear area of blank sample/Peak area of standard×100
System suitability:
System suitability is based on the concept that all the analytical operations, procedures, equipment and samples to be analyzed are an integral part of the system for the intended use. The system suitability experiment was performed by injecting the hesperetin sample (six replicates) into HPLC. Retention times and peak areas of samples were recorded, and Relative Standard Deviation (RSD) (%) was calculated.
Linearity:
Linearity is the ability of the developed method for a particular range to obtain responses proportional to the amount of analyte present in the sample. To determine the linearity of hesperetin, eight different concentrations over a range of 25 to 2500 ng/mL were injected and analyzed using the developed HPLC method. Peak areas were recorded for all the samples, and the calibration curve was plotted for concentrations (x-axis) against average peak areas (y-axis). Linear regression analysis was performed to determine the coefficient of determinate (r2), slope, and intercept.
Precision:
Precision measures the degree of scatter (closeness of agreement) between a series of data values from multiple sampling of the same homogenous samples and analyzed under the same analytical conditions[11]. Precision was investigated as intraday and inter-day precision at three different concentration levels, LQC (75 ng/mL), MQC (1250 ng/mL), and HQC (2000 ng/mL). The peak area was recorded for all the samples, and RSD (%) was calculated.
Accuracy:
The accuracy of an analytical method expresses the closeness of the experimental value to the true value[11]. Accuracy of the analytical method was performed at three different concentration levels, 75 % (937.5 ng/mL), 100 % (1250 ng/mL), and 125 % (1562.5 ng/mL). Set I samples (937.5, 1250, and 1562.5 ng/mL) were prepared directly from stock solution (1000 μg/mL), and Set II samples (937.5, 1250, and 1562.5 ng/mL) were prepared by allegations method using sample concentration of 250 ng/ml and 2500 ng/mL. All the samples were analyzed in triplicates using the developed HPLC method, and data was recorded.
LOD and LOQ:
The LOD is the least concentration that can be detected with the developed method, and the limit of quantification LOQ is the least concentration that can be quantified. LOD and LOQ were calculated from formulas (equations 2 and 3).
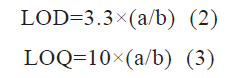
Where a is the standard deviation of the y-intercept and b is the slope of the calibration curve
Robustness:
The robustness is the ability of an analytical method to remain unaffected by deliberate and small changes in the method parameters. In the current study, small, deliberate changes were applied for various method parameters such as injection volume, detection wavelength, column oven temperature, flow rate, the ratio of mobile phase composition and pH. Peak area, NTP and TF were recorded, and RSD (%) was calculated.
Applicability of the method:
Preparation of topical gel of hesperetin: The hesperetin-loaded gel was prepared following the previously reported method[15]. In brief, the gel base (carbopol approximately 1 % w/w) was prepared by soaking an accurately weighed amount of carbopol in milli-Q water for about 2 h to swell up. This swollen mixture was then kept under a mechanical stirrer at room temperature to achieve a uniform and clear homogenous dispersion of the mixture. The pH of the gel was adjusted to 7.4 using triethanolamine. An accurately weighed amount of hesperetin was added to the gel to achieve strength of 0.5 % w/w. Methylparaben and propylparaben were added as preservatives to the gel. The entire mixture was mechanically stirred to get a clear and homogenous gel and stored in an airtight container till further use.
Determination of drug content:
Accurately 0.1 g of gel was weighed and added 10 ml of HPLC grade methanol. This solution was sonicated for 15 min in a bath sonicator to extract the drug from the gel, followed by centrifugation at 10 000 rpm for 5 min. The supernatant solution was further diluted to get a final concentration of 1 μg/ml. Finally, the sample was transferred to an autosampler vial and analyzed on HPLC using a validated analytical method.
Statistical analysis:
All the results are expressed as mean±standard deviation. Linearity was calculated by linear regression analysis. All the statistical calculations were performed with GraphPad Prism software version 5.00 (San Diego, California, USA).
Results and Discussion
The development of a good chromatographic method involves the selection and optimization of various parameters such as stationary phase, mobile phase, and flow rate[16]. Selection of the stationary phase involves the selection of column chemistry, column dimensions, particle size and carbon loading. Silica gel is the most popular base material used in HPLC columns. It consists of Si atoms bridged by oxygen atoms, and on the surface, it carries OH groups (silanol groups). Surface modification of these silanol groups helps in obtaining specific properties to the column. Base Deactivated Silica (BDS) and Octadecylsilyl (ODS) are two different types of columns used in reverse-phase chromatography. The OH groups are blocked in BDS, whereas these functional groups are free and reactive in ODS type of columns. The shape of the column material, such as fully porous, core-shell, monolithic phase, and bound phase, are some of the varieties of particles. Fully porous particles are one of the most common particles used in HPLC with a particle size of 3 or 5 μm. Smaller particle size yields better resolution, but produces high back pressure compared to larger particles. Particle size distribution is also important as the wide particle size distribution is not suitable for good separation. Selection of the suitable pore size of the column packing is important during the method development. Analysis of large molecules requires column packing with larger pore size. For smaller molecules, pore size up to 120 Å is generally used[17].
Mobile phase selection is the other critical step in chromatographic method development. The mobile phase generally consists of a combination of aqueous and organic solutions. As pH is the most important variable in reverse phase chromatography of ionizable compounds, buffer solutions are used as the aqueous mobile phase to control the pH. The pH of the buffer solution to be used is decided based on the pka of the molecule to be analyzed. When pH is two units above the pKa of the acid, it will be >99 % ionized, and if the pH is two units below its pka it exists in an unionized form. Similarly, bases exist in the non-ionized form above their pka and ionized below their pka. Ionized and unionized forms are retained in a different manner in reverse-phase chromatography. Unionized form will be more hydrophobic and retains strongly. Thus, acids are retained more with mobile phase consisting of lower pH, whereas bases retain with high pH mobile phase. When pH of the mobile phase is near to pka, it affects not only the stability of the retention times but also the selectivity of the method when similar structures are present in the sample. Hence, to achieve a robust separation and good selectivity, pH of the mobile phase should be maintained at least two units above or below its pka [18]. In this current study, pH of the buffer was adjusted to 3.0 with orthophosphoric acid, which is lower than pka of hesperetin (7.92)[19].
Organic modifiers such as methanol and acetonitrile are the most commonly used solvents in reversephase chromatography. The aqueous component of the mobile phase usually exhibits less elution strength whereas less polar organic solvents show more elution strength. Hence a relative proportion of aqueous and organic solvents is required for good separation and to control the retention of the analyte on the column. Miscibility of the organic solvent with an aqueous portion of the mobile phase, viscosity, and safety are some parameters to be considered while selecting the organic modifier. For example, though isopropanol can provide high elution strength, its usage is limited because of its viscosity, which results in high back pressure. Acetonitrile and methanol are the most commonly used organic modifiers in preparing HPLC mobile phases. In the current study, acetonitrile was used as the organic modifier in the preparation of the mobile phase. The viscosity of the acetonitrile/water mixture is lower than that of methanol/water. Generally, lower back pressure is observed with an acetonitrile/water mixture than with methanol/water, which helps in saving column life. Moreover, the elution strength of acetonitrile is more therefore, shorter retention of analyte is observed in comparison to methanol[20]. Arya et al.[21] developed an HPLC method for the quantification of hesperetin along bicalutamide, where the CN column was used as the stationary phase. It was reported that the column was selected based on the affinity of the bicalutamide with the stationary phase and considering the high affinity of hesperetin towards the mobile phase (acetonitrile and acetic acid). The retention time of hesperetin was reported as 4.98 min with 1.0 ml/min[21]. In another study, Kanaze et al.[10] used C18 column to quantify hesperetin and naringenin and achieved good separation. And in this study, a mixture of methanol and acetic acid was used as the mobile phase at a flow rate of 1.0 ml/min to retain hesperetin at 16.1 min. In the current study, C18 ODS column was used with a mixture of acetonitrile and acidified water (in a ratio of 50:50, % v/v) as the mobile phase. Hesperetin was retained at 6.0 min with a mobile phase flow rate of 1.20 ml/min. As the total run time of the method is 10 min, it will be helpful in saving resources and also, fast analysis is possible with good retention.
Excipients present in samples may interfere with the analyte of our interest, which can cause misleading of actual sample data. Specificity experiment reveals the presence of any interference at the retention time of the analyte due to the various possible components in the samples[22]. In the current study, no interference was observed in the mobile phase injection at the analyte's retention time, which shows the specificity of the method for the quantification of hesperetin. A specificity experiment was also performed with the blank gel formulation and no interference was observed at the retention time of the analyte. The chromatogram of hesperetin and the mobile phase is depicted in fig. 1.

Fig. 1: Chromatogram comparison of mobile phase with hesperetin peak
In the system suitability experiment, RSD (%) was found to be <1 for retention time and peak area of hesperetin standard. The data evidence that all components of the instrument and set parameters are proper for the intended use of the method. System suitability data is presented in Table 1.
| S No | RT (min) | Peak area (mV-min) |
|---|---|---|
| 1 | 5.79 | 163324 |
| 2 | 5.79 | 162592 |
| 3 | 5.78 | 162572 |
| 4 | 5.78 | 162238 |
| 5 | 5.78 | 162929 |
| 6 | 5.79 | 161197 |
| Mean±SD | 5.78±0.004 | 162475.30±726.57 |
| RSD (%) | 0.07 | 0.45 |
Note: The mean was calculated using 6 replicates (n=6) for each parameter, RT: Retention Time; SD: Standard Deviation; RSD: Relative Standard Deviation
Table 1: System Suitability Data of Hesperetin
The responses were found to be linear over a concentration range of 25 to 2500 ng/ml. The calibration curve and linear regression equation are presented in fig. 2. The data represents a good correlation between peak area and concentration level of hesperetin with a coefficient of determinate of 0.9999.

Fig. 2: Calibration curve of hesperetin
RSD (%) of intraday and inter-day precision determined with three concentration levels was found to be <1 and <2, respectively (data is presented in Table 2). The data proves that the developed method is precise for the quantitative determination of hesperetin in analytical samples.
| S No | Intraday precision peak area (mV-min) | Interday precision peak area (mV-min) | ||||
|---|---|---|---|---|---|---|
| LQC (75 ng/ml) | MQC (1250 ng/ml) | HQC (2000 ng/ml) | LQC (75 ng/ml) | MQC (1250 ng/ml) | HQC (2000 ng/ml) | |
| 1 | 4520 | 80812 | 126640 | 4520 | 80812 | 126640 |
| 2 | 4569 | 81628 | 127128 | 4569 | 81628 | 127128 |
| 3 | 4596 | 81526 | 128303 | 4596 | 81526 | 128303 |
| 4 | 4517 | 81557 | 127562 | 4517 | 81557 | 127562 |
| 5 | 4531 | 81134 | 126686 | 4531 | 81134 | 126686 |
| 6 | 4523 | 81416 | 126468 | 4523 | 81416 | 126468 |
| 7 | 4490 | 83046 | 126787 | 4504 | 81932 | 125023 |
| 8 | 4545 | 82210 | 127269 | 4504 | 81486 | 125442 |
| 9 | 4470 | 82390 | 127498 | 4538 | 82096 | 125533 |
| 10 | 4510 | 82322 | 127387 | 4407 | 81806 | 125241 |
| 11 | 4468 | 82017 | 127068 | 4437 | 81534 | 124812 |
| 12 | 4462 | 81874 | 127064 | 4418 | 81073 | 124606 |
| Mean± SD | 4516.75±40.88 | 81827.66±611.66 | 127155±502.47 | 4505.33±57.71 | 81500.00±363.55 | 126120.30±1181.53 |
| RSD (%) | 0.91 | 0.75 | 0.4 | 1.28 | 0.45 | 0.94 |
Note: The mean was calculated using 12 replicates (n=12) for each experiment; RSD: relative standard deviation; SD: standard deviation
Table 2: Intraday and Inter Day Precision Data of Hesperetin
Mean recovery was calculated for all three levels (75, 100, and 125 %) to determine the accuracy through the recovery experiment. Percent mean recovery was calculated for all the samples, and data are presented in Table 3. The accuracy was found to be between 95-100 % for both Set I and Set II samples, indicating a good recovery of hesperetin and hence the accuracy of the method developed for its quantification.
| S No | Level | Concentration (ng/ml) | Accuracy (%) | |||
|---|---|---|---|---|---|---|
| Samples prepared by stock solution | Samples prepared by allegations method (250 and 2500 ng/ml) | |||||
| 1 | 75 % | 937.5 | 97.77±0.17 | 97.35-98.20 | 97.76±0.29 | 97.45-98.07 |
| 2 | 100 % | 1250 | 95.87±0.42 | 94.82-96.92 | 95.68±0.80 | 94.85-96.51 |
| 3 | 125 % | 1562.5 | 94.29±0.26 | 93.65-94.93 | 95.11±0.12 | 94.98-95.23 |
Note: The data presented as mean±SD, n=3, SD: Standard Deviation; CI: Confidence Interval
Table 3: Recovery Data of Hesperetin
LOD and LOQ were found to be 1.61 and 4.89 ng/ ml, respectively which is calculated from SD of the y-intercept and slope of the calibration curve. The lower LOD and LOQ values emphasize the high sensitivity of the method for detecting and quantifying hesperetin in analytical samples.
The robustness of the experiment was evaluated by analyzing samples after making slight deliberate changes in instrument parameters. Data of the robustness experiment is presented in Table 4, and from the data, it is evident that the developed method remains unaffected with small changes made in the method parameters.
| S No | Parameter | RT | Peak area (mV-min) | NTP (USP) (per column length) | TF (5%) | |
|---|---|---|---|---|---|---|
| 1 | IV (µL)-01 | 18 | 6.01±0.00 | 74505.50±245.80 | 6487.33±29.97 | 1.13±0.00 |
| 2 | IV (µL)-02 | 22 | 6.01±0.01 | 91004.50±213.35 | 65072.00±25.20 | 1.13±0.00 |
| 3 | DW (nm)-01 | 286 | 5.96±0.01 | 81433.00±214.64 | 6825.50±45.95 | 1.11±0.00 |
| 4 | DW (nm)-02 | 290 | 5.96±0.01 | 80676.34±379.41 | 6831.33±48.07 | 1.10±0.00 |
| 5 | CVT (°C)-01 | 22 | 6.12±0.00 | 82241.34±246.96 | 6727.17±50.02 | 1.10±0.00 |
| 6 | CVT (°C)-02 | 28 | 5.81±0.00 | 82210.84±113.76 | 6964.17±18.95 | 1.10±0.00 |
| 7 | FR-01 | 1.1 | 6.46±0.00 | 88543.16±206.70 | 6733.67±40.72 | 1.11±0.00 |
| 8 | FR-02 | 1.3 | 5.48±0.00 | 75589.80±379.61 | 6686.40±55.66 | 1.10±0.00 |
| 9 | MP ratio-01 | 48:52:00 | 6.18±0.01 | 81625.16±301.45 | 6914.33±62.59 | 1.10±0.00 |
| 10 | MP ratio-02 | 52:48:00 | 5.33±0.01 | 81969.16±212.32 | 6628.50±32.01 | 1.10±0.00 |
| 11 | Buffer pH-01 | 2.8 | 5.64±0.03 | 82831.00±222.25 | 6744.33±28.73 | 1.12±0.00 |
| 12 | Buffer pH-02 | 3.2 | 5.62±0.00 | 82361.84±334.09 | 6788.17±49.00 | 1.11±0.00 |
Note IV: Injection Volume; DW: Detector Wavelength; CVT: Column Oven Temperature; FR: Flow Rate in ml/min; MP ratio: Mobile Phase ratio (acetonitrile: OPA, pH 3.0); NTP: Number of Theoretical Plates; RT: Retention Time; SD: Standard Deviation, The data presented as mean±SD, n=6
Table 4: Robustness Results of Hesperetin
Hesperetin-loaded carbopol gel was prepared by a simple method using a mechanical stirrer. As a part of the gel characterization, drug content was determined using a validated HPLC analytical method. Hesperetin was extracted from the gel using methanol as the extraction solvent. Bath sonication of gel dissolved in methanol solution further assisted in the complete extraction of the drug. The drug content of the formulation was found to be 98.46±0.42 % (n= 6). The data evidence the complete extraction of the drug from the formulation. It also evidences the reliability of the analytical method used for quantifying hesperetin from the formulation.
In conclusion, the study shows that the HPLC method developed for the quantification of hesperetin was found to be sensitive, fast, accurate, precise, robust and economical. Method validation data supports that the developed HPLC method is within the acceptance criteria of the guidelines. The method followed for extracting hesperetin from topical gel formulation is capable of extracting the drug from the formulation. Further, the validated HPLC method was useful in the quantification of extracted hesperetin from the topical gel formulation. Finally, from this study, we can conclude that the HPLC method developed will be useful for the routine quantification of hesperetin in analytical samples with good sensitivity and accuracy.
Acknowledgements:
The authors are thankful to the Indian Council of Medical Research (ICMR), New Delhi, India, for providing financial assistance to Srinivas Reddy Jitta in the form of a Senior Research Fellowship (SRF) (project Ref. no. 45/73/2018-Nan/BMS, dated June 4, 2019). The authors are thankful to the Department of Pharmaceutics of Manipal College of Pharmaceutical Sciences (MCOPS) and Manipal Academy of Higher Education (MAHE), Manipal, Karnataka- 576 104, for providing the conveniences to carry out the research work.
Author contributions:
S. R. Jitta and Arunima Chauhan contributed equally to this work
Conflict of interest:
The authors declared no conflict of interests.
References
- Chen YJ, Kong L, Tang ZZ, Zhang YM, Liu Y, Wang TY, et al. Hesperetin ameliorates diabetic nephropathy in rats by activating Nrf2/ARE/glyoxalase 1 pathway. Biomed Pharmacother 2019;111:1166-75.
- Wang Y, Liu S, Dong W, Qu X, Huang C, Yan T, et al. Combination of hesperetin and platinum enhances anticancer effect on lung adenocarcinoma. Biomed Pharmacother 2019;113:108779.
[Crossref] [Google Scholar] [PubMed]
- Jayaraman R, Subramani S, Abdullah SH, Udaiyar M. Antihyperglycemic effect of hesperetin, a citrus flavonoid, extenuates hyperglycemia and exploring the potential role in antioxidant and antihyperlipidemic in streptozotocin-induced diabetic rats. Biomed Pharmacother 2018;97:98-106.
[Crossref] [Google Scholar] [PubMed]
- Hesperetin. National library of medicine; PubChem 2021.
- Valipour J, Mojaverrostami S, Abouhamzeh B, Abdollahi M. Protective effects of hesperetin on the quality of sperm, apoptosis, lipid peroxidation, and oxidative stress during the process of cryopreservation: An experimental study. Int J Reprod BioMed 2021;19(1):35-46.
[Crossref] [Google Scholar] [PubMed]
- Sohel M, Sultana H, Sultana T, Al Amin M, Aktar S, Ali MC, et al. Chemotherapeutic potential of hesperetin for cancer treatment, with mechanistic insights: A comprehensive review. Heliyon 2022;8(1):e08815.
[Crossref] [Google Scholar] [PubMed]
- Kumar M, Dahiya V, Kasala ER, Bodduluru LN, Lahkar M. The renoprotective activity of hesperetin in cisplatin induced nephrotoxicity in rats: Molecular and biochemical evidence. Biomed Pharmacother 2017;89:1207-15.
[Crossref] [Google Scholar] [PubMed]
- Famurewa AC, Renu K, Eladl MA, Chakraborty R, Myakala H, El-Sherbiny M, et al. Hesperidin and hesperetin against heavy metal toxicity: Insight on the molecular mechanism of mitigation. Biomed Pharmacother 2022;149:112914.
[Crossref] [Google Scholar] [PubMed]
- Evans JA, Mendonca P, Soliman KF. Neuroprotective effects and therapeutic potential of the citrus flavonoid hesperetin in neurodegenerative diseases. Nutrients 2022;14(11):2228.
[Crossref] [Google Scholar] [PubMed]
- Kanaze FI, Kokkalou E, Georgarakis M, Niopas I. A validated solid-phase extraction HPLC method for the simultaneous determination of the citrus flavanone aglycones hesperetin and naringenin in urine. J Pharm Biomed Anal 2004;36(1):175-81.
[Crossref] [Google Scholar] [PubMed]
- Saifullah M, McCullum R, Vuong Q. Maximising extraction yields of gallic acid and hesperetin from lemon myrtle (Backhousia citriodora) leaf using microwave assisted extraction. Results Chem 2020;2:100080.
- Wang W, Zhao L, Huang H, Yao J, Zhou L, Wang D, et al. Development of an ultra-high performance liquid chromatography method for simultaneous determination of six active compounds in Fructus aurantii and rat plasma and its application to a comparative pharmacokinetic study in rats administered with different doses. J Anal Methods Chem 2018;2018:7579136.
[Crossref] [Google Scholar] [PubMed]
- FDA GR. Validation of chromatographic methods. center for drug evaluation and research (CDER). Food and Drug Administration. 1994:2.
- International Conference on Harmonization. Validation of analytical procedures: Text and methodology Q2 (R1). In: International Conference on Harmonization 2005.
- Vaz VM, Jitta SR, Verma R, Kumar L. Hesperetin loaded proposomal gel for topical antioxidant activity. J Drug Deliv Sci Technol 2021;66:102873.
- Joshi VS, Kumar V, Rathore AS. Role of organic modifier and gradient shape in RP-HPLC separation: analysis of GCSF variants. J Chromatogr Sci 2015;53(3):417-23.
[Crossref] [Google Scholar] [PubMed]
- Selection of a suitable HPLC column. Analytics Shop 2021.
- A Guide to HPLC and LC-MS buffer selection. ACE HPLC Columns. 2021.
- Hesperetin. Drug bank online. 2021.
- Trevor Hopkins on behalf of advanced chromatography technologies. The role of methanol and acetonitrile as organic modifiers in reversed-phase liquid chromatography. Chromatography Today 2021.
- Arya A, Khandelwal K, Singh A, Ahmad H, Agrawal S, Khatik R, et al. Validation of RP-HPLC method for simultaneous quantification of bicalutamide and hesperetin in polycaprolactone-bicalutamide-hesperetin-chitosan nanoparticles. J Chromatogr Sci 2015;53(9):1485-90.
[Crossref] [Google Scholar] [PubMed]
- Rao NM, Sankar DG. Development and validation of stability indicating RP-HPLC method for the estimation of cinacalcet hydrochloride in bulk and their formulations. Biointerface Res Appl Chem 2020;10(6):6610-8.





