- *Corresponding Author:
- S. Kumar
Product Development and Research, Sun Pharmaceutical Industries Limited, Sector-18, Gurgaon-122 015, India
E-mail: msk.vats@gmail.com
| Date of Submission | 30 November 2016 |
| Date of Revision | 11 February 2017 |
| Date of Acceptance | 20 July 2017 |
| Indian J Pharm Sci 2017;79(5):740-750 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
The main aim of the research work was to investigate the type and level of the polymer for solubility enhancement of nisoldipine by solid dispersion approach, followed by study of the formulation factors affecting design of a single unit, solid dispersion containing osmotic controlled release tablet. Solid dispersions with copovidone and Soluplus® were manufactured by hot melt extrusion technique, which offered possibility of scale up and commercialization. Dynamic vapour sorption and differential scanning calorimetry were used to characterize solid dispersions. Based on the glass transition temperature and dynamic vapour sorption data, nisoldipine-Soluplus®, at 1:1 ratio was selected to make dissolution enhanced solid dispersion system. The formulation factors affecting the drug release from the osmotic tablets such as type of dispersant, level of osmogen, %plasticizer in film coat and %coating, were studied by full factorial design. The drug release was found to increase with the increase in sodium chloride concentration in the core and polyethylene glycol level in the coating. The coating weight gain was found to decrease the drug release. Scanning electron microscopy revealed that the drug release occurred through the drilled hole and not through the extended release coating membrane. An interesting observation was found with respect to the dispersant type in the core. The high viscosity dispersant (Polyox) was found to facilitate complete release from the tablets compared to highly soluble, low viscosity dispersant (lactose). A design space was generated indicating that 36% osmogen in the core, a %polyethylene glycol level of 10-15% and weight gain at 5-7% can provide for a single core osmotic dosage form releasing ~80% drug in 12 h. The drug release from the monolithic tablets followed the Korsenmeyer-Peppas kinetics.
Keywords
Hot melt extrusion, dynamic vapour sorption, glass transition temperature, design of experiments, monolithic osmotic
The absorption and bioavailability of the poorly soluble drugs is rate limited by its solubility and dissolution, more so for Biopharmaceutics Classification System (BCS-II) drugs where the drug permeability is not a concern. Extended release dosage forms containing such poorly soluble drugs provide incomplete drug release and leads to variability in bioavailability [1]. Solid dispersion (SD) approach is widely used to address the poor solubility, but stability of amorphous form upon storage remains a concern. A decreased solubility and dissolution rate, due to the conversion from the amorphous (metastable) to the crystalline state during storage remains a challenge for amorphous SDs [2]. Therefore, the identification and understanding of the factors influencing crystallization from the amorphous state is one of the most important aspects of designing such systems. A controlled drug delivery system for SD systems is difficult to design, particularly in a reservoir based drug delivery system.
Push pull osmotic drug delivery systems (PPOP) were developed and commercialized for poorly water-soluble drugs like nifedipine (Procardia XL) and glipizide (Glucotrol XL). A sandwiched system for nifedipine, to avoid laser identification of the right side for drilling was also reported [3]. These systems are very sophisticated to manufacture and often involves multiple complex machinery e.g. solvent handling capable rapid mixer granulator, bilayer press and laser drilling machine. Monolithic osmotic systems offer a relatively simple design to address these issues. However, encasing of a poorly soluble drug in a reservoir would limit the surface area available for vehicular contact and would limit the drug release as per Noyes-Whitney equation [4]. Therefore, the solubility and dissolution profile of a poorly soluble drug must be altered to facilitate the drug release. A monolithic osmotic system or a single core osmotic tablet (SCOT) based on SD offers this flexibility. Literature reports incorporation of solubility enhanced form into osmotic system for many drugs like 10-hydroxycamptothecan [5], nifedipine [6,7], glipizide [8], atenolol [9] and diltiazem [10]. However, the reports about polymer level and type screening through glass transition temperature (Tg) approach and statistical evaluation of factors affecting design of monolithic osmotic dosage forms were scarce. The aim of present research work is to investigate the type and level of the polymer for solubility enhancement of nisoldipine by SD approach, followed by study of the formulation factors affecting design of a single unit, SD containing osmotic controlled release tablet. The present research work discusses about an alternate method to select the polymer type and drug load based on Differential scanning calorimetric (DSC) and dynamic vapour sorption (DVS) characterization followed by the use of factorial design in optimizing formulation factors affecting drug release from single core tablets containing poorly soluble drug in solubility enhanced form and ascertains the drug release mechanism from the osmotic tablet through SEM and dissolution data modeling.
Materials and Methods
Nisoldipine was procured from Erregierre, Italy. Inactive ingredients were sourced from Macron Inc., USA (sodium chloride fine grade, magnesium stearate), DFE Pharma, USA (lactose DCL 11), Clariant Inc., USA (polyethylene glycol, PEG 3350), Dow Chemicals (Polyox N-80), Eastman Inc., USA (cellulose acetate 398-10), BASF Corporation, USA (Soluplus® (SOLU), Kollidon VA64), Colorcon, India (Opadry® pink 03B540127). The materials were obtained from the Indian suppliers. All other chemicals were of analytical grade and were used as obtained. Nisoldipine is prone to photolytic degradation and hence all the experiments were carried out using golden fluorescent light and analysis was carried out using low actinic amber colour glassware.
Phase solubility studies
Solubility measurements were performed in triplicate using the method reported by Higuchi and Connors [11]. An excess amount of nisoldipine was added to purified water containing increasing concentrations (0-10% W/V) of SOLU and copovidone (COP). The vials were sealed and shaken at 37±0.5° for 72 h in a thermostatically controlled orbital shaker cum incubator (Colton, India) and the samples were filtered through a 0.45 μ polyvinylidene fluoride (PVDF) filter. The filtrate was suitably diluted and the concentration in the solution was determined spectrophotometrically at λmax 238 nm (UV-2450, Shimadzu, Japan). The measurements were performed in triplicate.
Preparation of amorphous nisoldipine
Amorphous nisoldipine was prepared by method reported in literature [12]. A stainless-steel beaker containing ~5 g drug was heated in an oven and held at 200° for 5 min. It was then cooled by immersion into liquid nitrogen. The quench-cooled drug was ground gently with a mortar and pestle and screened using a #60 sieve. This was stored at 0° over phosphorous pentoxide till further use.
Preparation of SD by hot melt extrusion
Hot melt extrusion process offers an advantage of being a solvent free process for SD preparation and has excellent scalability [13]. For SD preparation using the hot melt extrusion technique, the drug and polymer were mixed geometrically and the blend was fed into extrusion chamber of a co-rotating twin screw hot-melt extruder (Pharm11, Thermo Fisher Scientific, Germany) at a constant feed rate of 50 rpm and a powder feed rate of 200 g/h. The length to diameter ratio was 40 and the temperature of seven heating zones starting from hopper side towards the die (2 mm round) were set at 110, 130, 140, 150, 150, 160 and 160° (for SOLU based) and 110, 130, 150, 150, 170, 180 and 180° (for COP based). The extruded thread was subsequently passed through an in line milling unit. The milled extrudes were cooled to room temperature and pulverized and screened via 425 μ sieve (#40 ASTM).
Preparation of physical mixtures
Physical mixtures were prepared by mixing the components using a glass mortar and pestle for 5 min. The samples were then dried at 40° under vacuum for 3 h. Dried physical mixtures were used immediately in the experiments.
Determination of density
The density of amorphous (quench-cooled) nisoldipine and melt cooled polymers were measured by helium pycnometry (Quantachrome Corp., India) at room temperature. All the measurements were made in triplicate.
DSC studies
For DSC studies, the samples (~10 mg) were sealed in perforated aluminium pans and thermo grams were obtained at heating rate of 10°/min in the temperature range of 30°-200° in the atmosphere of nitrogen (Perkin Elmer, DSC 8000, Japan) to determine the Tg. The measurements were performed in triplicate and the thermograms were detected and analysed by Pyris software.
DVS studies
To evaluate the moisture susceptibility of the drug, polymers, physical mixtures and SDs, DVS studies were performed [14]. A known amount of sample was weighed and kept in the sample holder in a controlled temperature and pressure chamber (DVS advantage, surface measurement systems, UK). The change in mass was monitored with varying humidity conditions to understand the effect of moisture. All the samples were then analysed “as is” at a constant temperature of 30° and programmed for sorption-desorption at 0%, 75% and 0% for a period of 6 h.
Full factorial (24) design of experiments (DoE)
The selected SD formulation was formulated into a monolithic/single core osmotic system. Based on the literature and initial risk assessment, a 24 full factorial DoE was selected to evaluate the factors affecting drug release from an oral osmotic delivery system (Table 1).
| Formulation variables | Role | Level 1 | Level 2 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Factors | ||||||||||||||
| Osmogen in core (X1) | Continuous | 15 | 45 | |||||||||||
| Dispersant in core (X2) | Categorical | Lactose | Polyox | |||||||||||
| %Plasticizer in the film coat (X3) | Continuous | 10 | 30 | |||||||||||
| %coating weight gain (X4) | Continuous | 5 | 10 | |||||||||||
| Responses | Constraints | |||||||||||||
| %Cumulative drug release at 2 h (Y1) | 0%≤ Y≤30% | |||||||||||||
| %Cumulative drug release at 4 h (Y2) | 40%≤Y2≤70% | |||||||||||||
| %Cumulative drug release at 8 h (Y3) | 60%≤Y3≤80% | |||||||||||||
| %Cumulative drug release at 12 h (Y4) | Maximize (>80%) | |||||||||||||
| Experimental design | Responses | |||||||||||||
| Code | Pattern | Variables | (%Cumulative drug release ±SD, n=3) | |||||||||||
| X1 | X2 | X3 | X4 | Y1 (2 h) | Y2 (4 h) | Y3 (8 h) | Y4 (12 h) | |||||||
| OS1 | +−−+ | 15 | 10 | 5 | DCL-11 | 13.83±2.14 | 28.17±1.94 | 52.67±1.97 | 78.17±2.56 | |||||
| OS 2 | +−++ | 15 | 10 | 5 | N-80 | 9.75±0.78 | 39.5±3.54 | 67.5±2.12 | 94.5±0.35 | |||||
| OS 3 | +−+− | 15 | 10 | 10 | DCL-11 | 7.75±2.22 | 17.25±4.57 | 44.75±1.71 | 66.25±3.20 | |||||
| OS 4 | ++−− | 15 | 10 | 10 | N-80 | 0.25±0.50 | 15.25±1.26 | 44.5±3.11 | 82±1.63 | |||||
| OS 5 | ++++ | 15 | 30 | 5 | DCL-11 | 43.5±2.89 | 65.25±2.06 | 78.75±1.61 | 85.25±2.06 | |||||
| OS 6 | −−−+ | 15 | 30 | 5 | N-80 | 38±0.82 | 72.5±0.58 | 88.25±2.06 | 97.75±0.50 | |||||
| OS 7 | −+−+ | 15 | 30 | 10 | DCL-11 | 32.75±0.50 | 54.5±2.08 | 68.25±1.68 | 74.75±1.71 | |||||
| OS 8 | +−−− | 15 | 30 | 10 | N-80 | 28±3.27 | 58.67±3.06 | 78.67±3.68 | 83±3.61 | |||||
| OS 9 | −+++ | 45 | 10 | 5 | DCL-11 | 25.33±2.08 | 57.33±2.52 | 66.67±0.58 | 89.67±4.73 | |||||
| OS 10 | −+−− | 45 | 10 | 5 | N-80 | 25.33±0.58 | 50.32±2.54 | 76.67±2.08 | 94.67±0.58 | |||||
| OS 11 | −−++ | 45 | 10 | 10 | DCL-11 | 20±1.83 | 47.25±2.22 | 66±2.16 | 74.5±1.29 | |||||
| OS 12 | −−−− | 45 | 10 | 10 | N-80 | 27.25±3.77 | 67.33±4.59 | 80.28±2.78 | 88.25±1.89 | |||||
| OS 13 | ++−+ | 45 | 30 | 5 | DCL-11 | 70.53±2.39 | 93.88±1.14 | 98.25±2.63 | 98.75±2.06 | |||||
| OS 14 | +++− | 45 | 30 | 5 | N-80 | 79.5±1.91 | 101.25±0.50 | 100.75±1.26 | 100±0.82 | |||||
| OS 15 | −−+− | 45 | 30 | 10 | DCL-11 | 57.83±2.72 | 79.5±2.89 | 87.75±1.69 | 92.75±0.96 | |||||
| OS 16 | −++− | 45 | 30 | 10 | N-80 | 59.6±1.69 | 80.4±2.51 | 95.51±1.26 | 100.75±0.97 | |||||
Table 1: Factors, levels, constaints and responses for formulation of monolithic osmotic tablet
Manufacturing of monolithic osmotic core tablets
Selected SD (powdered) and dispersant (lactose or Polyox) were screened through #30 sieve and mixed. Sodium chloride was further screened and mixed with the SD-dispersant mixture, geometrically, for 5 min. Magnesium stearate screened through #60 sieve was added to the blend and it was lubricated for 5 min. The composition had SD and lubricant as fixed part per tablet at 34 mg and 10 mg, respectively and the dispersant quantity was varied as per the experimental design to maintain a tablet weight of 200 mg. The blends were compressed into tablet using 8.0 mm standard round concave punch on a tablet press (Cadmach, India) and tablet thickness was maintained at 4.0 to 4.5 mm. Each tablet contained 17 mg equivalent nisoldipine.
Extended release coating
The extended release coating solution was prepared by dissolving cellulose acetate in acetone under stirring (Remi, India) till clear solution was formed. PEG was dissolved in water and added slowly to the cellulose acetate solution and stirred for at least 60 min before use. The solution concentration in all the experiments was 5% w/w. The core tablets were loaded into a coating pan fitted with 1.0 mm nozzle (Gansons, India) and coated using their respective coating solutions, to the %coating required as per experimental design. The process parameters were inlet temperature: 35- 42°, exhaust temperature: 25-28°, atomization air: 1.0- 1.5 bar, pan speed: 4-8 rpm, spray rate: 5-12 g/min. Post coating, the tablets were dried in the pan itself at increased product temperature of ~35-40° for about 1 h. The tablets were drilled with a single hole of 0.5×0.5 mm, using a dentist’s drill (SMT Microtech, India) and cured in a vacuum oven (Pharmalab, India) for 24 h at 45°.
Top film coating
The drug is photosensitive and hence the extended release coated tablets were coated with an additional top colour coating to provide light protection. The drilled extended release tablets were loaded into a coating pan fitted with 1.0 mm nozzle (Gansons, India) and coated with an aqueous dispersion of Opadry® pink (10% w/w). The dispersion was filtered through #40 before coating on the tablets. The process parameters were inlet temperature: 45-50°, exhaust temperature: 38-45°, atomization air: 1.5-2.0 bar, pan speed: 4-8 rpm, spray rate: 5-10 g/min. After ~5% w/w coating, the tablets were dried in the pan itself at increased product temperature of ~40-45° for about 30 min. The top colour coated tablets were stored in triple laminated bags till further use.
Drug content estimation
The tablets were subjected to drug content determination by dissolving in methanol under sonication for 2 h. The supernatant was filtered and diluted with 0.1 N HCl containing 0.25% sodium lauryl sulphate (SLS) to approx. 17 μg/ml. The absorbance of these dilutions was measured at 238 nm and read from the corresponding standard curve equation (UV-2450, Shimadzu, Japan).
Dissolution studies
The in vitro dissolution behaviour of SCOTs was studied using dissolution system equipped with auto sampler (Distek Inc., USA). The dissolution studies were performed in USP Dissolution apparatus type II (paddle) in 900 ml of 0.1 N HCl containing 0.25% SLS as dissolution media at 50 rpm and 37±0.5° (n=3). The dissolution test was performed for 12 h with 5 ml sampling at 2, 4, 8, 12 h and replaced with same volume of fresh media post each sampling. The samples were filtered using 0.45 μ PVDF filter, diluted and analysed by UV spectrophotometer at 238 nm. Cumulative amount of drug dissolved (with sampled volume adjustment) was calculated using calibration equation.
Scanning electron microscopy (SEM)
The surface of the extended release coated tablets of the optimized formulation (OS17) before and after dissolution was studied using SEM (Jeol-JSM-5300, Japan). For after dissolution sample, the tablets were carefully removed after 12 h from dissolution media, wiped with a tissue paper and dried in an oven for 6 h at 45°. The samples were mounted on a glass stub with double-sided carbon tape and coated under vacuum with gold in an argon atmosphere prior to observation.
Statistical analysis of the 24 factorial response data
JMP® software (version 12, SAS Inc., USA) was used for the evaluation of the statistical experimental design. Means were compared by ANOVA. The level of significance was set at α=0.05. Suitable regression models were generated and response surface methodology was used to search for an optimized formula [15].
Evaluation of drug release kinetics
The in vitro drug release data of the optimized formulation (OS17) was analysed with various mathematical models (Korsmeyer-Peppas, first order, zero order) to ascertain the drug release kinetics using DD Solver, an MS Excel add in software package [16].
Results and Discussion
Nisoldipine belongs to BCS class II drugs. Its aqueous solubility was determined to be 5.91 μg/ml. The saturation solubility of drug was evaluated in SOLU solutions at 0-10% w/w concentration. The phase solubility curve has shown in Figure 1. The solubility of nisoldipine increased as a function of polymer concentration. The increase in solubility with COP (polyvinyl pyrrolidone with 40% vinyl acetate) can be attributed to a drug-polymer interaction owing to vinyl acetate moieties facilitating solubilisation. An AL-type phase solubility curve was seen (linear model, y=20.947x+6.6696, r²=0.987). The increase in solubility in presence of SOLU was even higher than COP. The data was modelled into a linear trend line (y=29.249+17.32, r²=0.988) and this too followed an AL type phase solubility curve. The drastic increase in solubility with the increased SOLU concentration can be reasoned basis the chemical nature of the polymer. SOLU has an amphiphilic molecular structure that acts as a polymeric solubilizer. Its large number of hydroxyl groups facilitates solubilisation by molecular interaction. Additionally, the polymer dissolves to form micelle structure, which facilitates the solubility enhancement [17]. This indicates that the use of higher quantity of polymer in SD will improve the dissolution in microenvironment, which can result in complete drug release from the formulation.

Figure 1: Nisoldipine phase solubility studies
Copovidone ( ) and Soluplus® (
) and Soluplus® ( ) (mean±SD, n=3)
) (mean±SD, n=3)
The amorphous form of drug has a higher solubility compared to the crystalline form and hence SD containing amorphous nisoldipine would facilitate the increase in dissolution rate. The DSC studies of pure nisoldipine indicated an endothermic event occurring between 148°-158° and exhibited a sharp melting point (MP) at 154°. To study the effect of level of drug loading on the Tg was investigated with COP and SOLU SDs using DSC. A typical DSC indicating Tg have shown in Figure 2A and B. The Tg of SOLU was found to be ~70° while that of COP ~102°. For SDs, only one Tg was observed meaning that none of the component was present in solitary state in significant quantities. At a low drug loading, little change was seen in the Tg. The theoretical values of Tg were predicted from of a modified form of the Gordon-Taylor Eqn. 1 [18,19]. Tg12=(w1Tg1+ Kw2Tg2)/(w1+Kw2), where Tg and w represent the glass transition temperature and weight fraction of the component 1 (drug) and component 2 (polymer), respectively. The value of K was calculated using Eqn. 2. K=d1Tg1/d2Tg2, where d are the density values. The density was determined by gas pycnometry and found to be 1.09, 1.22, 1.56 for quench cooled nisoldipine and melt cooled COP and SOLU, respectively. As seen in Figure 2C, the positive deviation observed, compared to the predicted data indicates stronger drug polymer interaction, either in number or strength compared to that between individual components [20]. The positive deviation observed is stronger for SOLU compared to COP and at 50% drug load, a Tg of 78° and 70° was observed respectively which is well above the intended storage temperature for SDs. Therefore, at this polymer level, recrystallization of amorphous dispersion may be prevented on storage at accelerated stability conditions as per International Conference on Harmonisation (ICH) guidelines [21].

Figure 2: Typical DSC images of solid dispersions showing Tg
(A) Nisoldipine-copovidone (6:1), Tg=56.5°, and (B) nisoldipine-Soluplus (3:1), Tg=59.6°. (C) Glass transition temperature (Tg)
detected in copovidone ( ); Soluplus (
); Soluplus ( ) solid dispersions, as a function of concentration of polymers. Dotted lines are predictions
for fitted to equations 1 and 2. Error bars indicate mean±SD, n=3
) solid dispersions, as a function of concentration of polymers. Dotted lines are predictions
for fitted to equations 1 and 2. Error bars indicate mean±SD, n=3
Presence of moisture can reduce the Tg and may facilitate the crystallization of the drug from SD [22]. The change in mass upon exposure to 75% RH is depicted in Figure 3. The maximum moisture uptake was ~9% for pure COP. The moisture sorption of physical mixtures with NIS-COP increased with an increase in polymer content. However, for the corresponding SDs, the moisture uptake was significantly less (P<0.05). The change in mass rose sharply after 25% polymer load. This can be reasoned due to the hydrogen bonding between COP and drug thus the potential water binding sites are occupied at 1:1 level. The freeing up of the interaction sites in the polymer chains after this level caused increased moisture uptake. The reduction in moisture absorption level in dispersions compared to the physical mixtures has also been reported for albendazole-PVP systems [23].

Figure 3: Dynamic vapour sorption plot of solid dispersions
Copovidone () and Soluplus (), dotted lines are plots for
respective physical mixtures
For nisoldipine-SOLU system, the increase in moisture uptake was not significant since SOLU is a non- hygroscopic material. However, the SOLU-physical mixture showed comparatively more moisture uptake than SD. SOLU SD also showed sharp rise in moisture uptake beyond 50% polymer, which might be due to freeing up of hydrogen bonding sites for water binding.
Nisoldipine-SOLU was selected to form SD at 1:1 level since the phase solubility data indicated that increase in the solubility per unit polymer was higher than COP and the Tg was found to be ~68°, which is well above the desired storage temperature. Moreover, SOLU based SD was not significantly affected by moisture up to 50% level. Further, this SD system was used for formulating into the SCOT.
Osmotic drug delivery system can provide for a near zero order drug release and is one of the most prominent reservoir drug delivery system [24]. The components of a typical single core osmotic system include the active part (drug or SD), a dispersant, and lubricant with a semi-permeable membrane (SPM) coating comprising of cellulose acetate and a plasticizer like PEG. An osmogen (e.g. sodium chloride) is often added to the core to facilitate water influx through the SPM. Based on the literature and initial risk assessment, a fullfactorial design was selected. During compression, it was difficult to compress the blends of OS9 to OS16 into tablets. This could be reasoned because of the very high sodium chloride level. The dissolution data from the formulations was provided in the Table 1. A mathematical model was generated to correlate the factors to predict response and the actual by predicted plots Figure 4 showed an r2 value between 0.95 to 0.99, and P<0.05. This means that the prediction models were significant. Percent PEG and %NaCl level in the core were found to be significant (P<0.05) factor affecting the initial dissolution at 2 h. The magnitude of effect was %PEG>%NaCl>%weight gain. An interaction between NaCl-PEG and NaCl filler was also found to be significant. This means that osmogen level in the core plays a significant role in water influx through the semi permeable membrane which affects the drug release [25]. At 4 h also, %PEG and %NaCl level in the core were found to be significant (P<0.05) factor affecting the dissolution at 4 h. No interaction between the factors was found to be significant. This means that apart from the osmogen level in the core, the %PEG level too plays a significant role in determining water influx through the SPM, which affects the drug release. At 8 h and 12 h, all the four factors studied were found to be significant (P<0.05) factor affecting the dissolution. However, no interaction between them was found to be significant. To envisage the effect of the factors studied on the response, a generalized prediction profiler was generated (Figure 5). The prediction profiler indicated that the level of sodium chloride in the core affects the drug release at all the time points. The osmogen helps imbibe water into the core and generate an osmotic pressure that facilitates the drug release. The %PEG level in the coating determines the permeability of the SPM and it also impacts the drug release throughout the time points. The %coating weight gain has less affect compared to osmogen and %PEG level. A higher %coating hinders complete release from the dosage form.

Figure 4: Actual by predicted plots for responses
(A) Y1 predicted P=0.0001, RSq=0.99, RMSE=3.5584 (B) Y2 predicted P=0.0098, RSq=0.95, RMSE=9.3361 (C) Y3 predicted
P=0.0037, RSq=0.97, RMSE=5.3385 (D) Y4 predicted P=0.0023, RSq=0.97, RMSE=2.8745

Figure 5: Generalized prediction profiler showing change in response for the factors varied
The prediction profiler indicated that the level of sodium chloride in the core affects the drug release at all the time points. The
osmogen helps imbibe water into the core and generate an osmotic pressure that facilitates the drug release. The %PEG level in
the coating determines the permeability of the semi-permeable membrane and it also impacts the drug release throughout the
time points. The %coating weight gain has less affect compared to osmogen and %PEG level. A higher %coating hinders complete
release from the dosage form
An interesting observation was seen on the dispersant/ filler type. Compared to the lactose, which is highly water soluble material, the drug release, especially at the later time points was found to increase when dispersant was polyox. This can be explained by high viscosity of polyox compared to lactose, which disperse the drug to extrude out of the hole. It might be possible that rate of release of lactose, a very highly soluble material, be faster than that of the drug. Based on the above observations, a contour profiler was generated, to provide the drug release within the constraints listed in Table 2 and have shown in Figure 6A. The contour profiler, representing the design space, indicates that at approx. 36% osmogen in the core, a %PEG level of 10- 15% and 5-7% weight gain can provide for a osmotic dosage form releasing ~80% drug in 12 h.
| Coefficients | Y1 (h) | Y2 (h) | Y3 (h) | Y4 (h) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Coefficient | P-value | Coefficient | P-value | Coefficient | P-value | Coefficient | P-value | ||
| Intercept | 56.75 | <0.0001* | 78.25 | <0.0001* | 85.5 | <0.0001* | 87.75 | <0.0001* | |
| NaCl (15,45) | 23.48 | <0.0001* | 19.13 | 0.0118* | 12.75 | 0.0064* | 9.38 | 0.0017* | |
| %PEG (10,30) | 42.25 | <0.0001* | 33.01 | 0.0041* | 23.25 | 0.0016* | 10.75 | 0.0032* | |
| %Coating (5,10) | -7.44 | 0.0020* | -5.31 | 0.2459 | -2.13 | 0.4001 | -2.69 | 0.0833 | |
| Filler | -2.56 | 0.0991 | -2.56 | 0.5540 | -3.75 | 0.1657 | -2.44 | 0.1075 | |
| NaCl* %PEG | 13.31 | 0.0018* | -3.56 | 0.6326 | -1.88 | 0.6593 | 4.31 | 0.1019 | |
| NaCl* %Coating | -1.22 | 0.3172 | 2.91 | 0.4443 | 3.56 | 0.1353 | 1.97 | 0.1274 | |
| %PEG* %Coating | -5.63 | 0.0121* | -3.63 | 0.4725 | -1 | 0.7233 | 1.88 | 0.2488 | |
| NaCl* filler | -3.28 | 0.0304* | -0.09 | 0.9797 | 0 | 1.0000 | 2.34 | 0.0817 | |
| %PEG* filler | 0.13 | 0.9352 | 0.38 | 0.9391 | 1.25 | 0.6593 | 2.38 | 0.1593 | |
| %Coating* filler | -0.10 | 0.8079 | -0.19 | 0.9391 | 0.38 | 0.7900 | -0.44 | 0.5692 | |
| Model statistics | |||||||||
| R2 | 0.99 | 0.95 | 0.97 | 0.97 | |||||
| Adj, R2 | 0.98 | 0.81 | 0.93 | 0.92 | |||||
| Prob >F | <0.0001 | 0.0098 | 0.0037 | 0.0023 | |||||
| R2 | 0.99 | 0.95 | 0.97 | 0.97 | |||||
P-value is Prob>|t|, *indicates P<0.05, significant term
Table 2: Regression coefficients and level of significance for each response

Figure 6: Contour profiler showing design space for ER coating
Actual () and predicted (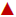 ). Dissolution profile (n=3,
mean±SD) from optimized osmotic tablet in 0.1 N HCl media
). Dissolution profile (n=3,
mean±SD) from optimized osmotic tablet in 0.1 N HCl media
Based on the prediction from DOE model, a batch was manufactured from within the design space (OS17) to confirm actual vs. predicted drug release, the composition of which is provided in Table 3. The actual versus predicted profile of the final formulation showed an F2 value (similarity factor) of 63 indicating a good correlation between the actual and predicted profile (Figure 6B). The drug content in optimized formulation was found to be 102.5±0.86% (n=3).
To investigate the drug release mechanism from the osmotic tablet containing SD, a scanning electron microscopic study was carried out. The extended release coating film before and after dissolution was studied (at ER stage) as shown in Figure 7. The drug release might have happened through the pores formed by dissolving of PEG present in the film or through the drilled hole. However, the surface view of the film on tablet surface after the dissolution did not show formation of any pores in the film. This indicates that the film had a semi-permeable characteristic, allowing only water to permeate through it and the drug release happened entirely by extrusion through the hole. The “ghost shell” at the end of the dissolution studies appeared somewhat swollen due to the hydrodynamic pressure created by osmosis. However, there was no crack formation or physical damage to the film at the selected plasticizer level.

Figure 7: Scanning electron microscope image of extended release
film on tablet
(A) Before dissolution, (B) after dissolution
Among the various mathematical models studied to understand the release kinetics, overall r2 and AIC value criteria [26] were used to select the most appropriate model. The parameters obtained were listed in Table 3. The drug release kinetics follows a Korsmeyer-Peppas model based on the lowest AIC value criteria. The n value indicates a non Fickian or Anomalous diffusion pattern.
| Composition | |
|---|---|
| Ingredients | mg/tab |
| Solid dispersion (1:1) | 34.00 |
| Sodium chloride | 72.00 |
| Polyethylene oxide | 84.00 |
| Magnesium stearate | 10.00 |
| Cellulose acetate | 7.40 |
| Polyethylene glycol | 3.60 |
| Acetone* | qs |
| Purified water* | qs |
| Opadry® Pink | 9.00 |
| Purified water* | qs |
| Total Tablet weight | 220.00 |
| Drug Release kinetics | |
| Korsmeyer-Peppas r2=0.99, KKP=16.24, AIC=10.90, n=0.64 | |
| First order r2=0.99, K1=0.13, AIC=13.62 | |
| Zero order r2=0.82, K0=7.19, AIC=24.84 | |
*Lost during processing, qs-quantity sufficient, AIC=Akaike information criteria
Table 3: Final formula and drug release kintics of monolithic osmotic tablets (os 17)
The drug delivery system design studied in current research work provides an insight into screening of the right polymer type and level through the Tg approach followed by design and optimization of monolithic reservoir based delivery system, uncommon for controlled release of poorly soluble drugs. Glass transition temperature and DVS studies guided the polymer type and level selection to predict the most suitable polymer to provide a thermodynamically stable SD. SOLU was selected to form SD by solvent less and industrially scalable hot-melt extrusion method at 50% drug load. Moreover, SOLU based SD was not significantly affected by moisture up to 50% level. Further, statistical DoE was extensively applied to investigate the factors affecting drug release from an osmotic tablet containing dissolution enhanced SD. A mathematical model and design space generated thereof indicated that at approx. 36% osmogen in the core, a %PEG level of 10-15% and 5-7% weight gain can provide for an osmotic dosage form releasing ~80% drug in 12 h. The final formulation showed a high degree of statistical similarity in the actual versus predicted dissolution profiles, indicating the prediction ability and validation of the design space. The in vivo performance of such dual mechanism systems (solubility enhanced and controlled release at the same time) can further be explored.
Acknowledgements
The authors would like to thank Sun Pharmaceutical Industries Limited for providing required facilities to carry out this research work.
Conflicts of interest
There are no conflicts of interest.
Financial support and sponsorship
Nil.
References
- Stegemann S, Leveiller F, Franchi D, De JH, Linden H. When poor solubility becomes an issue: from early stage to proof of concept. Eur J Pharm Sci 2007;31:249-61.
- Mooter GVD, Brande VD, Augustijns P, Kinget R. Glass forming properties of benzodiazepines and co-evaporate systems with poly(hydroxyethyl methacrylate). J Therm Anal Cal 1999;57:493-507.
- Liu L, Khang G, Rhee JM, Lee HB. Monolithic osmotic tablet system for nifedipine delivery. J Control Release 2000;67:309-22.
- Ho HO, Su HL, Tsai T, Sheu MT. The preparation and characterization of solid dispersions on pellets using a fluidized bed system. Int J Pharm 1996;139:223-29.
- Chen H, Jiang G, Ding F. Monolithic osmotic tablet containing solid dispersion of 10-hydroxycamptothecin. Drug Dev Ind Pharm 2009;35:131-37.
- Kanagale P, Patel V, Venkatesan N, Jain M, Patel P, Misra A. Pharmaceutical development of solid dispersion based osmotic drug delivery system for nifedipine. Curr Drug Del 2008;5:306-11.
- Liu L, Ku J, Khang G, Lee B, Rhee JM, Lee HB. Nifedipine controlled delivery by sandwiched osmotic tablet system. J Control Release 2000;68:145-56.
- Patel CG, Asodaria VK, Patel HP, Shah RD. Development of controlled release osmotic pump tablet of glipizide solid dispersion. Curr Drug Del 2014;11:817-27.
- Liu L, Wang X. Solubility-modulated monolithic osmotic pump tablet for atenolol delivery. Eur J Pharm Biopharm 2008;68:298-302.
- Prabakaran D, Singh P, Kanaujia P, Vyas SP. Effect of hydrophilic polymers on the release of diltiazem hydrochloride from elementary osmotic pumps. Int J Pharm 2003;259:173-79.
- Higuchi T, Connors KA. Phase solubility techniques. Adv Anal Chem Instr 1965;4:117-212.
- Kalaiselvan R, Mohanta G, Kannan K, Manna P, Manavalan R. Optimization of drug polymer mixing ratio in albendazole polyvinylpyrrolidone solid dispersion by moisture absorption studies. Acta Pharm Sci 2006;48:141-51.
- Karanth H, Shenoy VS, Murthy RR. Industrially feasible alternative approaches in the manufacture of solid dispersions: A technical report. AAPS PharmSci Tech 2006;7:31-8.
- Enose AA, Dasan PK, Sivaramakrishnan H, Shah SM. Formulation and characterization of solid dispersion prepared by hot melt mixing: a fast screening approach for polymer selection. J Pharm 2014;2014:1-13.
- Kim MS, Kim JS, You YH, Park HJ, Lee S, Park JS, et al. Development and optimization of a novel oral controlled delivery system for tamsulosin hydrochloride using response surface methodology. Int J Pharm 2007;341(1):97-104.
- Zhang Y, Huo M, Zhou J, Zou A, Li W, Yao C, et al. DDSolver: an add in program for modeling and comparison of drug dissolution profiles. AAPS J 2010;12:263-71.
- Zhang K, Yu H, Luo Q, Yang S, Lin X, Zhang Y, et al. Increased dissolution and oral absorption of itraconazole/soluplus extrudate compared with itraconazole nanosuspension. Eur J Pharm Biopharm 2013;85:1285-92.
- Gordon M, Taylor JS. Ideal copolymers and the second‐order transitions of synthetic rubbers. I. Non crystalline copolymers. J App Chem 1952;2:493-500.
- Sertsou G, Butler J, Hempenstall J, Rades T. Solvent change coprecipitation with hydroxypropyl methylcellulose phthalate to improve dissolution characteristics of a poorly water soluble drug. J Pharm Pharmacol 2002;54:1041-47.
- Schneider H, Rieger J, Penzel E. The glass transition temperature of random copolymers: 2. Extension of the Gordon Taylor equation for asymmetric Tg vs composition curves. Polymer 1997;38:1323-37.
- https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q1A_R2/Step4/Q1A_R2__Guideline.pdf.
- Qian F, Huang J, Zhu Q, Haddadin R, Gawel J, Garmise R, et al. Is a distinctive single Tg a reliable indicator for the homogeneity of amorphous solid dispersion? Int J Pharm 2010;395:232-35.
- Kalaiselvan R, Mohanta G, Manna P, Manavalan R. Inhibition of albendazole crystallization in poly (vinylpyrrolidone) solid molecular dispersions. Pharmazie 2006;61:618-24.
- Verma RK, Arora S, Garg S. Osmotic pumps in drug delivery. Crit Rev Ther Drug Carrier Syst 2004;21:477-520.
- Verma, RK, Kaushal AM, Garg S. Development and evaluation of extended release formulations of isosorbide mononitrate based on osmotic technology. Int J Pharm 2003;263:9-24.
- Akaike H. Information theory and an extension of the maximum likelihood principle. In: Kotzs S, editor. Breakthroughs in statistics. New York: Springer; 1998. p. 199-213.





