- *Corresponding Author:
- O. N. C. Umeh
Departments of Pharmaceutical Technology and Industrial Pharmacy, University of Nigeria, Nsukka-410 001, Enugu state, Nigeria
E-mail: ogochukwuonugha@yahoo.com
| Date of Submission | 11 February 2013 |
| Date of Revision | 01 October 2013 |
| Date of Acceptance | 05 October 2013 |
| Indian J Pharm Sci 2013; 75(6): 726-729 |
Abstract
Studies have shown that when compressing drugs with low aqueous solubility, the solubility of diluents selected is very crucial as it influences the disintegration, dissolution and bioavailability of such drugs. Based on these reports, the present study was undertaken to investigate the effect of some commonly used hydrophilic tablet diluents (lactose, sucrose, mannitol and dextrose) on the in vitro release properties of griseofulvin from compressed tablets. Griseofulvin granules and tablets were prepared using the wet granulation method. Tablet properties evaluated as a function of the diluents used include, hardness, friability, dissolution profile and dissolution efficiency at 60 min. Results obtained indicated variability in griseofulvin release in the presence of the diluents. The relative enhanced dissolution effects of the four hydrophilic diluents is in the order of dextrose>sucrose>lactose>mannitol. All the griseofulvin tablet batches produced exhibited a better drug release (in terms of rate and extent of release) than a commercially available tablet sample of griseofulvin (Fulcin®). The results of the dissolution efficiency (DE60min) are 91.7, 83.5, 48.7, 35.3 and 15.6% for dextrose, sucrose, lactose, mannitol and fulcin®, respectively. The overall results indicated that dextrose or sucrose can be utilised to improve the in vitro release profile and hence in vivo bioavailability of griseofulvin from compressed tablets.
Keywords
Griseofulvin, hydrophilic diluent, lactose, sucrose, mannitol, dextrose, dissolution efficiency
Potential bioavailability problems are prevalent with extremely hydrophobic drugs (aqueous solubility less than 0.1 mg/ml at 37°) due to erratic or incomplete drug absorption from the gastrointestinal (GIT) [1]. The rate limiting step in the process of drug absorption for such poorly soluble drugs is dissolution. In vitro dissolution is often applied to predict the in vivo product’s performance especially in cases of poorly soluble drugs and extended release formulations [2]. Studies have shown that when rapidly soluble (hydrophilic) substances are used as diluents (e.g. lactose and sucrose) in tablet formulations it leads to rapid disintegration and thus dissolution of the tablet [3].
Griseofulvin is a potent antibiotic fungistatic drug that inhibits fungal cell mitosis by causing disruption of the mitotic spindle structure, thereby arresting the metaphase of cell division [4,5]. It is administered orally in the treatment of dermatophyte and ringworm infections of the scalp, hair, nails and skin [4]. It is an essential drug in less developed countries and especially Nigeria where diseases and lack of elementary healthcare are the major medical concern. However, bioavailability problems due to solubility difficulties of griseofulvin have proved a set back to the use of the drug. For years, various efforts have been devised to combat this difficulty and to improve the dissolution profile of griseofulvin. Griseofulvin has been successfully mixed with nontoxic, water soluble polymers such as polyvinylpyrrolidone or hydroxypropyl cellulose and spray-dried before treatment with a wetting agent such as sodium lauryl sulphate (SLS) or benzalkonium chloride [6,7]. Size reduction of the powder molecules (ultramicronisation) [3,7] and variations in certain formulation methods has also been employed [3]. In the present work the comparative performance of four hydrophilic diluents (lactose, sucrose, mannitol and dextrose) on the release profile of griseofulvin tablets was investigated and compared with the release performance of a commercial sample, Fulcin®.
Griseofulvin® used in this study is a free gift from Evans medical PLC, Nigeria. Lactose, sucrose, mannitol and dextrose were obtained from EGA chemicals, Germany, while maize starch was obtained from May and Baker, England. All the other reagents used are of Analar grade.
Four batches of griseofulvin tablets each containing one of the hydrophilic diluents (lactose, sucrose, mannitol and dextrose) were prepared using the wet granulation method according to the composition presented in Table 1. Griseofulvin powder, maize starch and either of the hydophilic diluents (lactose, sucrose, mannitol and dextrose) were mixed in a specimen bottle for 10 min. A 5% w/w gelatin was dispersed in distilled water to produce a mucillage. The mucillage was incorporated into the powder mixture in a mortar, titurated using a pestle to form a homogenous damp mass and the damp mass was screened through a 1.7 mm stainless steel sieve. The screened granules were dried in an oven at 60° for 24 h, rescreened through a 1.00 mm stainless steel sieve and stored in well closed specimen amber coloured bottles.
| Ingredients | Batches | |||
|---|---|---|---|---|
| I | II | III | IV | |
| Griseofulvin powder (mg) | 100 | 100 | 100 | 100 |
| Maize starch (mg) | 50 | 50 | 50 | 50 |
| Gelatin (mg) | 25 | 25 | 25 | 25 |
| Lactose (mg) q.s.to 500mg | 320 | - | - | - |
| Sucrose (mg) q.s to 500mg | - | 320 | - | - |
| Mannitol (mg) q.s to 500mg | - | - | 320 | - |
| Dextrose (mg) q.s to 500mg | - | - | - | 320 |
| Magnesium stearate (mg) | 5 | 5 | 5 | 5 |
Table 1: formula for preparing four batches of griseofulvin tablets
Prior to compression, all four granule batches were lubricated with 1% magnesium stearate. Lubricated granules were compressed into tablets in a BMI Manesty single punch tabletting machine fitted with 9.5 mm flat faced punches. Compression pressure was maintained at 282.5 MPa and 60 tablets were produced for each batch. Tablet weights ranged from 475 to 546 mg. Prepared tablets were evaluated for hardness, friability and assay of active ingredients using standard methods [8,9].
The dissolution profile of griseofulvin in each tablet batch was assessed using the USP XXI apparatus 2 (paddle method). The Erweka DT dissolution apparatus fitted with a paddle that rotated at 100 rpm was used. The dissolution media consisted of deionised water containing 540 mg% SLS maintained at 37±1°. A tablet from each batch was placed in a basket (mesh size 325 mm) immersed and firmly suspended half way into the dissolution media. A 5 ml sampling at predetermined time intervals was followed by fresh equivalent volume replacement with the dissolution media maintained at the same temperature. Each withdrawn sample was analysed spectrophometrically at 296 nm using a UV 2102 spectrophometer. The concentration of griseofulvin released over time was calculated from the Beer’s calibration plot. The percentage amount of griseofulvin released for each batch was plotted against time. The dissolution profile data were fitted into the dissolution efficiency (DE) equation proposed by Khan and Rhodes [10] as shown in equation 1.
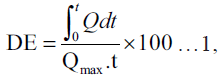 where Q is the percentage of
drug dissolved at time, t and Qmax is the maximum
amount of drug released. The time taken for 50%
of griseofulvin to be released from each batch was
estimated from the dissolution profile curve. The DEs
were calculated at t equal to 60 min. Values plotted
are a mean of two replicate determinations.
where Q is the percentage of
drug dissolved at time, t and Qmax is the maximum
amount of drug released. The time taken for 50%
of griseofulvin to be released from each batch was
estimated from the dissolution profile curve. The DEs
were calculated at t equal to 60 min. Values plotted
are a mean of two replicate determinations.
All values are expressed as mean±SD (standard deviation). Comparisons of all the data obtained from the dissolution study were made using one-way analysis of variance (ANOVA) with SPSS version 15. Statistical significance of the corresponding P value was tested at 5% confidence level (P<0.05) in all cases.
The presence of the diluents had variable effects on the hardness and friability of the tablets (Table 2). According to some published works, uncoated tablets with hardness of ≥4 kgf are considered adequate for handling and transportation [8,9]. From the results (Table 2), all the tablet batches are considered adequate in terms of hardness. The results for friability, also indicate that all the batches were within the acceptable limit of <1% [8,9]. The assay results of the batches ranged from 90.0 to 100.9% (Table 2). All the batches except the commercial sample (Fulcin®) had released up to 70% of griseofulvin within 45 min. There was a decrease in T50 (time taken for 50% of the drug to be released) as the DE increased. Batch D containing dextrose as diluent, had the highest DE while the commercial griseofulvin (Fulcin®) tablets had the least DE of 15.6%. A graphical representation of the dissolution profiles of the griseofulvin tablet batches are shown in fig. 1. Dextrose and sucrose enhanced the release of griseofulvin from the tablets than lactose and mannitol.
| Diluents | Parameters | ||||
|---|---|---|---|---|---|
| Hardness (kgf)* | Friability* | Assay (%)* | DE60 (%) | T50% (min) | |
| Lactose | 9.0 ± 1.9 | 0.12 ± 0.01 | 100.9 ± 0.00 | 48.7 | 29.30 |
| Sucrose | 4.2 ± 0.8 | 0.45 ± 0.03 | 106.0 ± 0.04 | 83.5 | 6.16 |
| Mannitol | 6.6 ± 1.6 | 0.15 ± 0.01 | 100.1 ± 0.00 | 35.3 | 45.14 |
| Dextrose | 5.8 ± 1.0 | 0.34 ± 0.02 | 99.3 ± 0.02 | 91.7 | 8.15 |
| Fulcin® | 5.4 ± 0.0 | 0.41 ± 0.03 | 90.0 ± 0.03 | 15.6 | 0.00 |
Table 2: some tablet properties of the griseofulvin batches

Figure 1: Release profiles of griseofulvin from the formulated and commercial Samples
The percentage concentration of griseofulvin released from the lactose (─♦─), sucrose (─■─), mannitol (─▲─) and dextrose (─×─) formulations were compared with that of fulcin® (─ж─), a commercially available sample. There was a significant increase (P<0.05) in the amount of griseofulvin released from the batches containing the four hydrophilic diluents than from the Fulcin® tablets.
Overall, there was a statistically significant increase (P<0.05) in the dissolution rate of griseofulvin in the presence of all the four hydrophilic diluents. It is evident from the release of griseofulvin from Fulcin® tablets, that the presence of SLS, a surfactant did not result in enhanced solubilisation and/or release of griseofulvin. For the test formulations, enhanced release of griseofulvin could be attributed to the combined effects of the hydrophilic diluents in the formulation and the surfactant (SLS) in the dissolution media. This result is consistent with earlier reports that SLS possesses good solubilising capacity at relatively low concentrations and could enhance the dissolution of poorly water soluble drugs [11-15]. Investigations have also demonstrated that the presence of surfactants influences tablet disintegration and produces finer dispersion of disintegrated particles with correspondingly larger surface area for drug dissolution [12-16]. Conflicting observations on the usefulness of surfactants such as SLS and PEG 4000 in enhancing GIT absorption of drugs have been reported [17,18]. Enhancement as well as inhibition of the GIT absorption and pharmacological activity of drugs have been observed when surfactants were added to drug formulations [17]. It has been stated that when the effect of incorporated surfactants in a formulation results in retardation of drug release, a special type of complexation reaction between the drug and surfactant may have taken place leading to aggregation of the surfactant molecules (micelles) [17,18]. Surfactant exert a two phase effect, which is concentration dependent. Below the critical micelle concentration (CMC), release/absorption of drugs may be enhanced due to a better contact of drug with the dissolution medium in vitro or absorbing membrane in vivo [19]. It has been observed that above the CMC, a portion of the drug molecules may become entrapped in micelles and as such, unavailable for absorption in vivo or release in vitro [19]. The release/absorption retarding effect usually predominate at higher surfactant concentrations because a larger fraction of the drug is bound to micelles [19]. The phenomenon of micellization at relatively high concentrations (540 mg%) might be considered responsible for the poor release of griseofulvin from the fulcin tablets.
The overall results indicated the usefulness of dextrose and sucrose as effective hydrophilic diluents that can be employed to improve the in vitro release profile and hence in vivo bioavailability of griseofulvin from compressed tablets. Lactose and mannitol may not be recommended as the first choice of diluents in the formulation of griseofulvin tablets.
References
- Muralidhar S, Devala G, Manohar B, Narayana TV, Ramesh R. Studiesto enhance dissolution properties of Celecoxib. PharmacieGlobale: IntJ Compr Pharm 2011;5:1-4.
- Bashir AA, Hatim SA, Ayman AK. Comparative dissolution ofdiltiazem immediate and extended release products using conventionalUSP and innovative dissolution paddles. Open Drug Deliv J2010;4:48-54.
- Kumar V, Alfonso RG. Dissolution. In: Remmington: The Science andPractice of Pharmacy, In: Alfonso RG, editor. Philadelphia: LippincottWiliams and Wilkins, 2005, p. 675-7.
- International Agency for Research on Cancer IARC Monograph, vol.79.Griseofulvin, 1987. p. 291-315. Available from: http://monographs.iarc.fr/ENG/Monographs/vol79/mono79-12.pdf. [Last accessed on 2011Jun 12].
- Fulvicin P/G (Schering). In: PDR Physician’s desk reference. 43rd ed.Oradell (NJ): Medical Economics Company; 1989, p. 1929.
- Finholt P, Salvang S. Dissolution kinetics of drugs in human gastricjuice: The role of surface tension. J Pharm Sci 1968;57:1322-6.
- Fell JT, Calvert RT, Rileybenthan P. Bioavailability of griseofulvinfrom a novel capsule formation. J Pharm Pharmacol 1978;30:479-82.
- Ofoefule SI, editor. A Textbook of Pharmaceutical Technology andIndustrial Pharmacy. 2nd ed. Lagos (Nig): Samakin Enterprises; 2002.p. 57-66.
- British Pharmacopoeia. London: The Pharmaceutical Press, HerMajesty’s Stationary Office; 2004. Appendix X1118 A259, X11DA261, X11J A273-4.
- Khan CA, Rhodes CT. The concept of dissolution efficiency. J PharmPharmacol 1975;27:48-9.
- Welling PG, Lasagna L, Banakar UV. Increasing Efficiency and Costeffectiveness.In, Banakar UV, Makoid MC. editors, PharmaceuticalIssues in The Drug Development Process. New York: Marcell Dekker;1996, p. 117-68.
- Crison JR, Weiner ND, Amidon GL. Dissolution media for in vitrotesting of water-insoluble drugs: Effect of surfactant purity andelectrolyte on in vitro dissolution of carbamazepine in aqueoussolutions of sodium laurylsulfate. J Pharm Sci 1997;86:384-8.
- Qazi S, Peter-Samuel NK, Venkatachalam TK, Uckun FM. Evaluatingdissolution profiles of an antiHIV agent using ANOVA and non-linearregression models in JMP software. Int J Pharm 2003;252:27-39.
- Shah VP, Konecny JJ, Everett RL, McCullough B, Noorizadeh AC,Skelly JP. In vitro dissolution profile of water-insoluble drug dosageforms in the presence of surfactants. J Pharm Res 1989;6:612-8.
- Sun W, Larive CK, Southard MZ. A mechanistic study of danazoldissolution in ionic surfactant solutions. J Pharm Sci 2003;92:424-35.
- Razzak SM, Khan F, Hossain M, Anika T, Moon SA. Impact ofsodium laurylsulphate on the release of carbamazepine from methocelK15M CR based matrix tablets. Bangladesh Pharm J 2012;15:79-82.
- Levy G. Biopharmaceutical considerations in dosage form design andevaluation. In, Sprowls JR, editor. Prescription Pharmacy, Philadelphia:JB Lippincott Co.; 1970, p. 72-3.
- Umeh ONC, Emeje MO, Ofoefule SI. Effect of some channelingagents on the release of DFK from a hydrophobic polymer matrix. ResJ Pharm BiolChemSci 2012;3:1174-7.
- Ofoefule SI, Chukwu A. Effects of polyethylene glycol 4000 andsodium laurylsulphate (SLS) on the release of hydrochlorthiazideembedded in dika fat matrix. Acta Pharm 2001;8:233-9.





