- *Corresponding Author:
- Benjaporn Buranrat
Faculty of Medicine,
Mahasarakham University,
Mueang,
Maha Sarakham 44000
Thailand
E-mail: buranrat@gmail.com
| Date of Received | 24 December 2020 |
| Date of Revision | 05 May 2021 |
| Date of Acceptance | 17 January 2022 |
| Indian J Pharm Sci 2022;84(1):72-79 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
Developing new treatment options for cholangiocarcinoma is necessary because of the adverse drug reactions and drug resistance problems associated with current chemotherapeutics. Therefore, this study examined cytotoxic, apoptotic and antimigratory effects of simvastatin used in combination with anticancer drugs (5-fluorouracil and cisplatin) against human cholangiocarcinoma cells and investigated the underlying molecular mechanism (s) of action. Proliferation, apoptosis and migration of cholangiocarcinoma cells were determined by sulforhodamine B, flow cytometry, colony formation, reactive oxygen species formation, caspase 3 activity, wound healing and western blotting. We observed that the two test statins, simvastatin and atorvastatin, enhanced 5-fluorouracil and cisplatin cytotoxicity against cholangiocarcinoma cells, simvastatin showed the higher effects than atorvastatin. Further, simvastatin plus 5-fluorouracil and cisplatin decreased colony formation and in combination induced p21 and reduced cyclin D1 protein. Furthermore, combination treatment augmented the apoptosis of cholangiocarcinoma cell through stimulating reactive oxygen species generation and caspase 3 activity as well as upregulating caspase 3 levels. Simvastatin also potentiated antimigratory effect of anticancer drugs via reduction in matrix metalloproteinase 9 levels. Accordingly, the combination of simvastatin with anticancer drugs could be considered a novel strategy to expand treatment options for cholangiocarcinoma.
Keywords
Cholangiocarcinoma, simvastatin, 5-fluorouracil, cisplatin, atorvastatin
Cholangiocarcinoma (CCA) or bile duct cancer is a deadly tumor that originates from both intrahepatic and extrahepatic epithelial bile duct cells[1,2]. The incidence of CCA is increasing worldwide and the northeast part of Thailand has the highest incidence in the world[3]. CCA is a devastating cancer and resistant to current anticancer drugs[4]. Treatment options are very limited and surgery is the only curative option for CCA patients. However, less than one-third of CCA patients are eligible for curative resection, resulting in poor prognosis[5,6]. Development of new therapeutic options is greatly needed and targeting the crucial pathway of cancer cells may be a valuable strategy for achieving efficacy and selective toxicity.
The Mevalonate (MVA) pathway plays a crucial role in cholesterol biosynthesis that has important functions in several cellular functions. Statins are competitive inhibitors of 3-Hydroxy-3-Methyl-Glutaryl-Coenzyme A (HMG-CoA) reductase, which is overexpressed in many cancer types[7,8]. Remarkably, statins have major effects on inhibition of cell proliferation, cell migration, cell invasion, cytoskeletal reorganization and cellular transformation[9] by interfering with Ras homologous (Rho) Guanosine Triphosphatase (GTPase) and other down-stream products of the MVA pathway. For apoptosis, a low concentration of simvastatin (5 μM) has been shown to induce CCA cell apoptosis within 6 h by inducing caspase 3/7 activity[10] and to lower phosphorylated Extracellular Signal-Regulated Kinase (p-ERK) expression[11]. Statins, in combination with anticancer drugs (gemcitabine, cisplatin, 5-Fluorouracil (5-FU)), have also been shown to induce significant CCA cell apoptosis. Moreover, statins suppress cancer cell migration by decreasing Matrix Metalloproteinase (MMP) 2 and MMP-9 expression[12]. Also, concurrent treatment with simvastatin and Nuclear Factor kappa B (NF-κB) inhibitor results in a synergistic suppression of Castration-Resistant Prostate Cancer (CRPC) cell growth[13].
Previously, we demonstrated the potent antiproliferative activities of simvastatin and atorvastatin on CCA cells[14] and Miller et al. showed that simvastatin stimulates CCA apoptosis by decreasing Ras-related C3 botulinum toxin substrate 1 (Rac1) enzyme activity[10]. However, limited information is available on the combined effects of statins and anticancer drugs on CCA. Therefore, this study evaluated the effects and mechanism of simvastatin in improving the sensitivity of CCA cells to the anticancer drugs, 5-FU and cisplatin.
Materials and Methods
Chemical reagents:
Dulbecco’s Modified Eagle’s Medium (DMEM), Fetal Bovine Serum (FBS) and other cell culture reagents were purchased from Gibco BRL Life Technologies (Grand Island, New York, United States of America (USA)). Simvastatin, atorvastatin, protease inhibitor cocktail, Dihydroethidium (DHE), Radioimmunoprecipitation (RIPA) lysis buffer and Sulforhodamine B (SRB) were obtained from Sigma-Aldrich (St. Louis, Missouri, USA). The primary antibodies against cyclin-dependent kinase inhibitor p21Cip/WAF1, caspase 3, beta (β)-actin and anti-rabbit Immunoglobulin G (IgG) Horseradish Peroxidase (HRP)-link antibody were purchased from Cell Signaling Technology (Beverly, Massachusetts, USA). Annexin V-Fluorescein Isothiocyanate (FITC) and Propidium Iodide (PI) dye solution were obtained from BD Biosciences (BD Biosciences, California, USA).
Cell culture and cytotoxicity:
The CCA cell line KKU-100, derived from a Thai patient, was obtained from the Department of Pathology at Khon Kaen University, Faculty of Medicine. KKU- 100 cells were maintained in DMEM with 10 % FBS, penicillin and streptomycin.
To assess cancer cell death, the SRB assay was used as described previously[14]. In brief, the cancer cells were exposed to 0-50 μM simvastatin and 0-50 μM atorvastatin with or without the anticancer drugs 5-FU 300 μM and cisplatin 2.5 μM for 24 h. Afterwards, cells were stained, solubilized and optical absorbance was measured at 540 nm using a spectrophotometer.
Colony formation:
To assess cancer cell regrowth, a colony formation assay was used as described previously[14]. In brief, cells were treated with 0-100 μM simvastatin, 0-50 μM 5-FU, 0-2.5 μM cisplatin and a combination of 10 μM simvastatin with or without 5 μM 5-FU and 0.25 μM cisplatin for 24 h. New and complete DMEM medium was then added and cells were cultured further for 15 d. Cells were stained with crystal violet, washed and the colonies were counted.
Wound healing:
To assess cancer cell migration, a wound healing assay was used[14]. Cells were scratched using a 0.2 ml sterile pipette tip. New complete DMEM medium was then added with 1 μM simvastatin, 300 M 5-FU, 0.1 μM cisplatin and a combination of 1 μM simvastatin with or without 300 μM 5-FU and 0.1 μM cisplatin. During the subsequent 48 h, plates were periodically imaged using an inverted microscope (CKX53, Olympus America, USA) with a 4× phase contrast objective. Distance of wound was measured by length of scratch.
Gelatin zymography:
To assess MMP-9 protein expression, the gelatin zymography assay was used[15]. Cells were treated with 1 μM simvastatin, 300 μM 5-FU, 0.1 μM cisplatin and a combination of 1 μM simvastatin with or without 300 μM 5-FU and 0.1 μM cisplatin for 48 h. The culture medium was harvested and protein concentration was measured. Protein was loaded onto 10 % Sodium Dodecyl Sulphate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) plus 0.01 % gelatin. The gels were washed with 2.5 % Triton X-100 and incubated in developing buffer overnight before staining with Coomassie brilliant blue R-250 to visualize band intensity.
Flow cytometry:
To assess cancer cell apoptosis, a flow cytometry assay was used. Cells were treated with 50 μM simvastatin, 300 μM 5-FU, 10 μM cisplatin and a combination of 50 μM simvastatin with or without 300 μM 5-FU and 10 μM cisplatin for 24 h. Next, cancer cells were collected and added to 100 μl binding assay buffer with 5 μl PE-Annexin V and 5 μl PI for 15 min prior to addition of 400 μl binding assay buffer. Cell apoptosis was assessed by flow cytometry (BD Biosciences, San Jose, California) using BD FACSDivaTM software.
Reactive Oxygen Species (ROS) formation:
To assess ROS formation, a DHE-fluorescent probe was used[14]. Cells were incubated with 50 μM simvastatin, 300 μM 5-FU, 10 μM cisplatin and a combination of 50 μM simvastatin with or without 300 μM 5-FU and 10 μM cisplatin plus 25 μM DHE for 90 min at 37°. Cells were then removed from the medium, washed and PBS buffer was added. The intensity of fluorescence was determined by a fluorescent microplate reader at 518 nm for excitation and 605 nm for emission.
Caspase 3 activity:
To assess cancer cell apoptosis, caspase 3 assay kits were used[15]. Cells were treated with 50 μM simvastatin, 300 μM 5-FU, 10 μM cisplatin and a combination of 50 μM simvastatin with or without 300 μM 5-FU and 10 μM cisplatin for 24 h, lysed with RIPA buffer and measured protein concentration. The protein (500 μg/ml) was mixed with assay buffer containing the substrate and incubated for 90 min. The fluorescence intensity was read at 360 nm for excitation and 460 nm for emission using a fluorescence microplate reader.
Protein expression:
To determine protein-related cell cycle and apoptosis expression levels, western blotting was used[15]. Cells were treated with 50 μM simvastatin, 300 M 5-FU, 10 μM cisplatin and a combination of 50 μM simvastatin with or without 300 μM 5-FU and 10 μM cisplatin for 24 h, lysed with RIPA buffer and protein concentration was measured. The protein (20 μg) was loaded onto 12 % SDS-PAGE, transferred onto Polyvinylidene Difluoride (PVDF) membranes and blocked with 2.5 % Bovine Serum Albumin (BSA). The membrane was exposed to primary antibodies, p21, cyclin D1, caspase 3 and β-actin (1:2500) overnight and incubated with secondary HRP-conjugated antibodies (1:5000) and visualized by ChemiDocTM Touch Imaging System.
Statistical analysis:
All data was represented as mean±Standard Error (SE) and tested for differences between control and treatment groups using the Student’s t-test and one-way Analysis of Variance (ANOVA), followed by Tukey’s post-hoc test. Statistical significance was taken as p<0.05.
Results and Discussion
Effects of statins with 5-FU and cisplatin on cell proliferation and cell apoptosis were shown below. The results found that simvastatin and atorvastatin (0-50 μM) inhibited CCA cell proliferation in a dose-dependent manner (fig. 1A). Simvastatin and atorvastatin in combination with the anticancer drugs, 5-FU and cisplatin inhibited proliferation more than statin treatment alone. The half-maximal inhibitory concentration (IC50) values in KKU -100 cells were 33.1±7.7 μM, 16.9±1.7 μM and 14.3 ±2.7 μM for simvastatin alone, simvastatin with 5-FU and simvastatin with cisplatin, respectively (fig. 1A).

Fig. 1: Effects of the statins, simvastatin and atorvastatin, in combination with the anticancer drugs, on proliferation and apoptosis
in KKU-100 cells. (A) Cell proliferation results obtained using the SRB assay for cells that had been treated with test compounds
for 24 h, for simvastatin (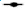 ) Sim; (
) Sim; (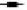 ) Sim+5-FU; (
) Sim+5-FU; (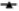 ) Sim+Cis; for atorvastatin (
) Sim+Cis; for atorvastatin (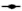 ) Ator; (
) Ator; ( ) Ator+5-FU; (
) Ator+5-FU; ( ) Ator+Cis
and (B) Apoptosis results generated by flow cytometry. Data are represented as mean±SE (n=3), *p<0.05 vs. simvastatin treatment
alone, #p<0.05 vs. atorvastatin treatment alone. Sim-simvastatin; Ator-atorvastatin; 5-FU-5-fluorouracil; Cis-cisplatin
) Ator+Cis
and (B) Apoptosis results generated by flow cytometry. Data are represented as mean±SE (n=3), *p<0.05 vs. simvastatin treatment
alone, #p<0.05 vs. atorvastatin treatment alone. Sim-simvastatin; Ator-atorvastatin; 5-FU-5-fluorouracil; Cis-cisplatin
Combination Index (CI) was adopted to evaluate whether combination treatment of simvastatin and both two cancer drugs had synergistic (CI<1), additive (CI=1) or antagonistic (CI>1) effects on CCA cells. Simvastatin (0-50 μM) plus cisplatin (2.5 μM) displayed the strongest synergy (CI<1) than 5-FU in combination treatment. In conclusion, simvastatin potentiated the effects of the anticancer drugs more than atorvastatin against the KKU-100 cell lines and we selected simvastatin for further study (Table 1).
| Simvastatin (mM) | Simvastatin+5-FU 300 mM (CI) | Simvastatin+cisplatin 2.5 mM (CI) | Atorvastatin (mM) | Atorvastatin+5-FU 300 mM (CI) | Atorvastatin+cisplatin 2.5 mM (CI) |
|---|---|---|---|---|---|
| 3.13 | 5.03 | 0.51 | 3.13 | 7.90 | 0.44 |
| 6.25 | 2.87 | 0.53 | 6.25 | 5.34 | 0.58 |
| 12.50 | 1.73 | 0.52 | 12.50 | 2.87 | 0.72 |
| 25.00 | 1.10 | 0.51 | 25.00 | 1.51 | 0.84 |
| 50.00 | 0.67 | 0.66 | 50.00 | 0.967 | 0.82 |
Note: *CI=1.00 additive effects; CI<1.00 synergistic effects; CI>1.00 antagonistic effects
Table 1: Combination Index Analysis of Simvastatin and Atorvastatin With 300 mM 5-FU and 2.5 mM Cisplatin
When apoptosis was studied using flow cytometry, simvastatin, 5-FU and cisplatin both caused reduction of cell proliferation and furthermore, augmented effects were demonstrated when used in different combinations, especially in simvastatin plus cisplatin (fig. 1B). Our results show that simvastatin enhances anticancer drug-induced CCA cell apoptosis.
Effects of simvastatin on 5-FU and cisplatin-induced inhibition of colony formation and the protein-related cell cycle in CCA cells were shown below. Simvastatin, 5-FU and cisplatin alone decreased colony forming ability of KKU-100 cells (fig. 2A), with IC50 values of 10.59±1.45 μM, 9.55±1.09 μM and 0.40±0.02 μM, respectively. Additionally, simvastatin in combination with either 5-FU or cisplatin enhanced the inhibitory effect on colony formation compared to treatment with the individual compounds (fig. 2A, p<0.05). This suggested that simvastatin potentiates the inhibitory activities of 5-FU and cisplatin.

Fig. 2: Effects of simvastatin and the anticancer drugs (5-FU and cisplatin) alone and in combination on cell regrowth and cell cyclerelated protein expression in KKU-100 cells. (A) Colony counts from cells cultured with test compounds for 14 d and (B) Protein expression as examined by western blotting. Data are represented as mean±SE (n=3), *p<0.05 vs. untreated control groups, #p<0.05 vs. simvastatin, †p<0.05 vs. anticancer drug. Sim-simvastatin; 5-FU-5-fluorouracil; Cis-Cisplatin
Furthermore, simvastatin and cisplatin were found to increase p21 levels in KKU-100 cells. The combination of simvastatin plus 5-FU increased p21 levels more than 5-FU treatment alone. Cisplatin with simvastatin protein expression results indicated that simvastatin did not upregulate p21 compared to the untreated control group. For cyclin D1 expression, simvastatin significantly decreased cyclin D1, but 5-FU and cisplatin did not. Cyclin D1 was also suppressed when simvastatin was combined with the two drugs (fig. 2B).
Effects of simvastatin on anticancer drug-induced ROS formation and caspase 3 activation were described below. Because ROS have been implicated in induction of cell death, we monitored the intracellular accumulation of ROS by the DHE-enhanced fluorescence method. Simvastatin and cisplatin, but not 5-FU, were found to induce significant intracellular ROS production (fig. 3A). Interestingly, simvastatin increased ROS formation in both the 5-FU and cisplatin groups. We also found that simvastatin and cisplatin treatment groups increased caspase 3 activity and that simvastatin in combination with the drugs further increased caspase 3 activity in a similar pattern to induction of ROS (fig. 3B). Caspase 3 protein levels found that caspase 3 protein was significantly induced by simvastatin treatment alone; however, 5-FU and cisplatin did not alter the caspase 3 levels. Interestingly, simvastatin potentiated anticancer drugs-induced caspase 3 levels. Our results show that simvastatin enhances anticancer drug-induced CCA cell death by increasing ROS formation and this is associated with increase in caspase 3 levels (fig. 3C and fig. 3D).

Fig. 3: Effects of simvastatin and the anticancer drugs alone and in combination on ROS formation, caspase 3 activity and caspase 3 protein expression. (A) ROS production quantified by the DHE-enhanced fluorescence method for cells incubated with compounds plus 25 μM DHE; (B) Caspase 3 activity and the lower images and charts; (C-D) Caspase 3 protein expression as examined by western blotting. Data are represented as mean±SE (n=3), *p<0.05 vs. control groups, #p<0.05 vs. simvastatin, †p<0.05 vs. anticancer drug. Sim-simvastatin; 5-FU-5-fluorouracil; Cis-Cisplatin
Effects of simvastatin on anticancer drug-induced suppression of cell migration were shown below.Simvastatin and 5-FU were found to significantly inhibit cancer cell migration. When simvastatin was combined with each of the two anticancer drugs, there was an additive effect and greater inhibition of KKU- 100 cell migration (fig. 4A). Simvastatin also reduced MMP-9 protein levels in the culture medium, but 5-FU and cisplatin did not (fig. 4B). In combination, we found that simvastatin plus cisplatin reduced MMP-9 levels more than simvastatin plus 5-FU. Our results show that simvastatin enhances the ability of anticancer drugs to inhibit CCA cell migration by reducing MMP- 9 protein levels.

Fig. 4: Effects of simvastatin and the anticancer drugs alone and in combination on cell migration and MMP 9 protein expression. (A) Cell migration results obtained using the wound healing assay and cells incubated with test compounds for 48 h; (B) Gelatin zymography data. Data are represented as mean±SE (n=3); *p<0.05 vs. control groups, #p<0.05 vs. simvastatin, †p<0.05 vs. anticancer drugs. Sim-simvastatin; 5-FU-5-fluorouracil; Cis-Cisplatin
This study has demonstrated that simvastatin inhibits proliferation, induces apoptosis and potentiates the effects of 5-FU and cisplatin. Simvastatin, in combination with anticancer drugs, can inhibit cancer cell proliferation by increasing p21, decreasing cyclin D1 levels and inducing apoptosis. These effects may be associated with induction of ROS formation and activation of caspase 3 activity and protein levels. Moreover, the combination treatments can suppress cancer cell migratory ability by decreasing MMP-9. Hence, simvastatin could be potentiating 5-FU and cisplatin activity against CCA cells.
In this study, simvastatin exhibited additive cytotoxic effects with the two anticancer drugs, 5-FU and cisplatin, to induce CCA cell death, as indicated by lowering of their median growth inhibitory concentrations. Simvastatin has previously been shown to induce Resting State/Growth 1 Phase (G0/G1) arrest by activating Adenosine Monophosphate- Activated Protein Kinase (AMPK) and inhibiting the Signal Transducer and Activator of Transcription 3-S phase kinase-associated protein 2 (STAT3-Skp2) axis, leading to upregulation of p21 and p27, the cell cycle dependent kinase inhibitors[16]. Amplification of cyclin D1 is frequently found in invasive breast carcinomas. In this study, simvastatin inhibited CCA cell proliferation by upregulating p21 and downregulating cyclin D1 protein expression and we found that simvastatin can potentiate 5-FU and cisplatin activity. As shown previously by Wang et al., showed that cerivastatin also significantly augments the cytotoxic effects of 5-FU against both drug-sensitive and drug-resistant cell lines by inhibiting NF-κB Deoxyribonucleic Acid (DNA) binding activity[17]. From our results, it indicated that 5-FU and cisplatin treatment alone did not alter the cyclin D1 and caspase 3 protein levels because the variation was detected in independent cancer cells and in this study we used the low concentration of these two anticancer drugs to combine the simvastatin. As shown by the study of Jin et al. cisplatin decreased the caspase 3 levels consistently which increased the cleaved caspase 3 level in lung cancer cells[18]. Moreover, cisplatin treatment alone caused induction of cyclin D1 levels as well[18]. In conclusion, statins and the anticancer drugs, 5-FU and cisplatin, act synergistically in inhibiting CCA cell proliferation.
Most cytotoxic anticancer drugs in current use induce apoptosis. Simvastatin has been shown previously to increase levels of ROS in Ocular Choroidal Melanoma-1 (OCM-1) cells and to trigger significant apoptosis, which was characterized by an increase in chromatin condensation and activation of caspase 9 and caspase 3[19]. Xing et al. have shown that the overproduction of ROS induces DNA damage, increases p53 levels and further activates cytochrome c-mediated caspase 3, leading to cell apoptosis[20]. Our study showed that simvastatin increases the CCA cell apoptosis caused by 5-FU and cisplatin by increasing ROS formation, inducing caspase 3 activity and also by increasing caspase 3 protein expression levels. In conclusion, statins together with the anticancer drugs, 5-FU and cisplatin, act synergistically to induce CCA cell apoptosis.
Metastasis is a basic characteristic of malignant cancer, where cells spread to many distant organs. It is a complex multistep process involving transformation of cells, changes in intercellular adhesion, migration and invasion[21]. Metastasis is an important problem that negatively affects prognosis in cancer patients[22,23]. Drugs which can block metastasis-associated steps could potentially be useful for cancer prevention and cancer treatment. In this study, treatment with simvastatin alone or in combination with 5-FU or cisplatin inhibited the migratory ability of CCA cells. This inhibition was associated with a down-regulation of MMP-9 activity, a degradation enzyme involved in cancer cell migration.
We found that, statins enhanced the cytotoxicity of two standard anticancer drugs, 5-FU and cisplatin, against CCA cells. This combination disrupted cell proliferation, increased apoptosis and decreased migration, possibly by interfering with growth and apoptosis-related cell proliferation. Based upon the results presented here, further studies are warranted to evaluate the therapeutic potential of oral statins in combination with anticancer drugs against CCA in experimental animal models. Ultimately, this combination could be a useful regimen for managing CCA.
Funding:
This work was financially supported by Mahasarakham University (Fast track 2020).
Acknowledgements:
The authors thank Dr. Adrian R. Plant and Professor Motoyuki Sumida (Mahasarakham University) for language-editing the manuscript.
Conflict of interests:
The authors declared no conflict of interest.
References
- Banales JM, Cardinale V, Carpino G, Marzioni M, Andersen JB, Invernizzi P, et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol 2016;13(5):261-80.
[Crossref] [Google Scholar] [PubMed]
- Khan SA, Taylor-Robinson SD, Toledano MB, Beck A, Elliott P, Thomas HC. Changing international trends in mortality rates for liver, biliary and pancreatic tumours. J Hepatol 2002;37(6):806-13.
[Crossref] [Google Scholar] [PubMed]
- Sripa B, Pairojkul C. Cholangiocarcinoma: Lessons from Thailand. Curr Opin Gastroenterol 2008;24(3):349-56.
[Crossref] [Google Scholar] [PubMed]
- Blendis L, Halpern Z. An increasing incidence of cholangiocarcinoma: Why? Gastroenterology 2004;127(3):1008-9.
- Farhat MH, Shamseddine AI, Tawil AN, Berjawi G, Sidani C, Shamseddeen W, et al. Prognostic factors in patients with advanced cholangiocarcinoma: Role of surgery, chemotherapy and body mass index. World J Gastroenterol 2008;14(20):3224-30.
[Crossref] [Google Scholar] [PubMed]
- Meza-Junco J, Montano-Loza AJ, Ma M, Wong W, Sawyer MB, Bain VG. Cholangiocarcinoma: Has there been any progress? Can J Gastroenterol 2010;24(1):52-7.
[Crossref] [Google Scholar] [PubMed]
- Altwairgi AK. Statins are potential anticancerous agents. Oncol Rep 2015;33(3):1019-39.
[Crossref] [Google Scholar] [PubMed]
- Matusewicz L, Meissner J, Toporkiewicz M, Sikorski AF. The effect of statins on cancer cells. Tumor Biol 2015;36(7):4889-904.
[Crossref] [Google Scholar] [PubMed]
- Jaffe AB, Hall A. Rho GTPases: Biochemistry and biology. Annu Rev Cell Dev Biol 2005;21:247-69.
[Crossref] [Google Scholar] [PubMed]
- Miller T, Yang F, Wise CE, Meng F, Priester S, Munshi MK, et al. Simvastatin stimulates apoptosis in cholangiocarcinoma by inhibition of Rac1 activity. Dig Liver Dis 2011;43(5):395-403.
[Crossref] [Google Scholar] [PubMed]
- Kamigaki M, Sasaki T, Serikawa M, Inoue M, Kobayashi K, Itsuki H, et al. Statins induce apoptosis and inhibit proliferation in cholangiocarcinoma cells. Int J Oncol 2011;39(3):561-8.
[Crossref] [Google Scholar] [PubMed]
- Gan J, Li P, Wang Z, Chen J, Liang X, Liu M, et al. Rosuvastatin suppresses platelet‑derived growth factor‑BB‑induced vascular smooth muscle cell proliferation and migration via the MAPK signaling pathway. Exp Ther Med 2013;6(4):899-903.
- Kang M, Lee KH, Lee HS, Jeong CW, Ku JH, Kim HH, et al. Concurrent treatment with simvastatin and NF-κB inhibitor in human castration-resistant prostate cancer cells exerts synergistic anti-cancer effects via control of the NF-κB/LIN28/let-7 miRNA signaling pathway. PLoS One 2017;12(9):e0184644.
[Crossref] [Google Scholar] [PubMed]
- Buranrat B, Senggunprai L, Prawan A, Kukongviriyapan V. Simvastatin and atorvastatin as inhibitors of proliferation and inducers of apoptosis in human cholangiocarcinoma cells. Life Sci 2016;153:41-9.
[Crossref] [Google Scholar] [PubMed]
- Buranrat B, Mairuae N, Konsue A. Cratoxy formosum leaf extract inhibits proliferation and migration of human breast cancer MCF-7 cells. Biomed Pharmacother 2017;90:77-84.
[Crossref] [Google Scholar] [PubMed]
- Wang ST, Ho HJ, Lin JT, Shieh JJ, Wu CY. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis 2017;8(2):e2626.
[Crossref] [Google Scholar] [PubMed]
- Wang W, Collie-Duguid E, Cassidy J. Cerivastatin enhances the cytotoxicity of 5-fluorouracil on chemosensitive and resistant colorectal cancer cell lines. FEBS Lett 2002;531(3):415-20.
[Crossref] [Google Scholar] [PubMed]
- Jin C, Song P, Pang J. The CK2 inhibitor CX4945 reverses cisplatin resistance in the A549/DDP human lung adenocarcinoma cell line. Oncol Lett 2019;18(4):3845-56.
- Wang Y, Xu SL, Wu YZ, Zhao MS, Xu WJ, Yang HY, et al. Simvastatin induces caspase-dependent apoptosis and activates P53 in OCM-1 cells. Exp Eye Res 2013;113:128-34.
[Crossref] [Google Scholar] [PubMed]
- Xing F, Zhan Q, He Y, Cui J, He S, Wang G. 1800MHz microwave induces p53 and p53-mediated caspase-3 activation leading to cell apoptosis in vitro. PLoS One 2016;11(9):e0163935.
[Crossref] [Google Scholar] [PubMed]
- Morrissey MA, Hagedorn EJ, Sherwood DR. Cell invasion through basement membrane: The netrin receptor DCC guides the way. Worm 2013;2(3):e26169.
[Crossref] [Google Scholar] [PubMed]
- Mehlen P, Puisieux A. Metastasis: A question of life or death. Nat Rev Cancer 2006;6(6):449-58.
[Crossref] [Google Scholar] [PubMed]
- Monteiro J, Fodde R. Cancer stemness and metastasis: Therapeutic consequences and perspectives. Eur J Cancer 2010;46(7):1198-203.
[Crossref] [Google Scholar] [PubMed]





