- *Corresponding Author:
- S. S. Rindhe
Department of Chemistry, New Arts Commerce and Science College, Ahmednagar- 414 001
E-mail: rindhe_ss@yahoo.com
| Date of Submission | 16 January 2009 |
| Date of Revision | 28 October 2009 |
| Date of Acceptance | 3 March 2010 |
| Indian J. Pharm. Sci., 2010, 72 (2): 231-235 |
Abstract
A series of substituted benzofuran derivatives were synthesized and characterized by spectral data. Some of the synthesized compounds were tested for in vitro antioxidant activity. Some of them have shown very good antioxidant activity. These compounds were also tested for antimicrobial activity against microbial strains viz. staphylococcus aureus (NCIM 5021) and salmonella typhimurium (NCIM 2501), but none of them showed any activity against these microorganisms.
Keywords
Benzofurans, antimicrobial activity, antioxidant activity
Various natural and synthetic benzofuran derivatives are found to possess diverse applications in the field of medicine. A crystalline antioxidant extracted from yeast was shown to protect in vitro the erythrocytes of vitamin E deficient rats from hemolysis. The structure of this compound was determined as benzofuran derivative [1]. It is well known that Vitamin E having chroman skeleton have a very good antioxidant activity. It has been reported that the activity is increased by the transformation of skeleton from chroman to benzofuran [2]. The antioxidant activity of a novel water soluble antioxidant of the benzofuran family (5-hydroxy-4,6,7-trimethyl-2,3- dihydrobenzofuran-2-acetic acid, BFA) is reported to possess better antioxidant activity than that of congener compound Trolox C [3].
Benzofuran in conjunction with cyclic β-amino hydroxamic acid scaffolds have found to possess very potent and selective tumor necrosis factor-ά converting enzyme (TACE) inhibitory activity. Similarly some of the benzofuran derivatives have shown very good antimicrobial activity and β-amyloid aggregation inhibitory activity [4-6]. Owing to the biological importance of benzofurans, we herein, report the synthesis and biological testing of some benzofurans.
In the present work, substituted phenacyl bromide (1) was treated with 2’-hydroxy-5’-nitro acetophenone (2) in presence of K2CO3 in DMF to get aryl-3- methyl-5-nitro-1-benzofuran-2-ylmethanone (3), which was further reduced to aryl-5-amino-3-methyl- 1-benzofuran-2-ylmethanone (4). Compound 4 was further treated with 5-chloronicotinoyl chloride to get N-(2-aroyl)-3-methyl-1-benzofuran-5-yl)-6- chloronicotinamide (5). Compound 5 was treated with various substituted amines in pyridine to get benzofuran derivatives 6(a-r), respectively.
All recorded melting points were determined in open capillary tubes and were uncorrected. IR spectra were recorded on Perkin-Elmer FTIR spectrophotometer in KBr disc. 1H NMR spectra were recorded on 400 MHz spectrophotometer in DMSO-d6 as a solvent and TMS as an internal standard. Peak values are shown in δ ppm. Mass spectra were obtained by Waters mass spectrometer.
General procedure employed for the preparation of aryl-3-methyl-5-nitro-1-benzofuran-2-ylmethanone (3) was equimolar mixture of substituted (0.1mol) phenacyl bromide 1 and (0.1 mol) 2’-hydroxy-5’- nitro-acetophenone 2 with (0.3 mol) K2CO3 in DMF was heated at 80° for 5 h. The reaction mixture was cooled to room temperature and then poured into ice cooled water. The solid product was separated by filtration and crystallized from DCM-hexane.
The procedure for the preparation of aryl-5-amino- 3-methyl-1-benzo furan-2-yl phenyl methanone (4) involved adding to a suspension of (0.1 mol) nitro derivative (3) in methanol (50 ml) 5 equivalents of SnCl2.2H2O and heating the reaction mixture at 60° for 4 h. The reaction mixture was cooled to room temperature. It was poured into liquid NH3 and filtered through hyflow. Filtrate was extracted by EtOAc. EtOAc layer separated, dried over Na2SO4 and concentrated under vacuum. The product was crystallized from ethanol.
N-(2-aroyl)-3-methyl-1-benzofuran-5-yl)-6-chloro nicotinamide (5) was prepared by stirring an equimolar mixture of (0.1 mol) amino compound (4) and (0.1 mol) 6-chloro nicotinoyl chloride in THF with (0.3 mol) K2CO3 for 6 h at room temperature. The reaction mixture was concentrated under vacuum and then poured into water. The solid product was separated by filtration and recrystallized from ethanol.
General procedure for the preparation of (6 a-r) involved adding a suspension of (0.1 mol) chloro compound 5 in pyridine to (0.12 mol) substituted amine. The suspension was heated for 12 h at 1100. The reaction mixture was then cooled to room temperature and poured into water. The solid product was separated by filtration and crystallized from DCM-Hexane. The compounds synthesized by the above procedure are listed in Table 1 and their structural data, spectral data and physical constant values are given in Tables 1 and 2.
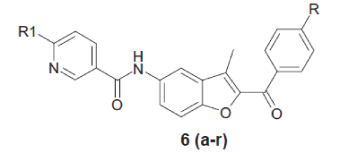
| Compds. | R1 | R | M.P. (0) | Yield (%) | Elemental analysis Calcd % (Found %) | |||
|---|---|---|---|---|---|---|---|---|
| C | H | N | ||||||
| 6a | morpholine | Br | 109-11 | 92 | 60.01 (60.00) | 4.26 (4.25) | 8.07 (8.05) | |
| 6b | OCH3 | 140-2 | 95 | 68.78 (68.77) | 5.34 (5.33) | 8.91 (8.90) | ||
| 6c | N-methyl piperazine | Br | 129-31 | 89 | 60.80 (60.79) | 4.72 (4.71) | 10.50 (10.49) | |
| 6d | OCH3 | 196-18 | 88 | 69.41 (69.40) | 5.82 (5.81) | 11.56 (11.55) | ||
| 6e | N-benzyl piperazine | Br | 118-20 | 95 | 65.03 (65.02) | 4.80 (4.79) | 9.19 (9.18) | |
| 6f | OCH3 | 119-21 | 90 | 72.84 (72.83) | 5.75 (5.74) | 9.99 (9.98) | ||
| 6g | thiomorpholine | Br | 147-9 | 59 | 58.21 (58.20) | 4.13 (4.12) | 7.83 (7.81) | |
| 6h | OCH3 | 135-7 | 55 | 66.51 (66.50) | 5.17 (5.17) | 8.62 (8.61) | ||
| 6i | pyrrolidine | Br | 128-30 | 69 | 61.91 (61.90) | 4.40 (4.39) | 8.33 (8.31) | |
| 6j | OCH3 | 126-8 | 67 | 71.19 (71.20) | 5.53 (5.52) | 9.22 (9.20) | ||
| 6k | piperidine | Br | 119-21 | 75 | 62.56 (62.55) | 4.67 (4.66) | 8.11 (8.10) | |
| 6l | OCH3 | 107-9 | 78 | 71.63 (71.62) | 5.80 (5.79) | 8.95 (8.94) | ||
| 6m | tetrahydroisoquinoline | Br | 131-3 | 88 | 65.73 (65.72) | 4.27 (4.28) | 7.42 (7.41) | |
| 6n | OCH3 | 139-41 | 80 | 74.26 (74.25) | 5.26 (5.25) | 8.12 (8.10) | ||
| 6o | 1-pyridyl-2-ylpiperazine | Br | 218-20 | 76 | 62.42 (64.41) | 4.39 (4.38) | 11.74 (11.73) | |
| 6p | OCH3 | 174-6 | 66 | 70.19 (70.18) | 5.34 (5.33) | 12.79 (12.77) | ||
| 6q | 1,4-dioxa-8-azaspiro[4,5] decane | Br | 137-9 | 60 | 60.43 (60.42) | 4.55 (4.54) | 7.29 (7.28) | |
| 6r | 2-piperzin-1-ylethanol | OCH3 | 102-4 | 55 | 67.69 (67.68) | 5.88 (5.87) | 10.89 (10.88) | |
Table 1: Structural and analytical data of the synthesized benzofuran derivatives
| Compd. | IR (cm-1) | Spectral data 1H NMR δ (ppm) | Mass M+ |
|---|---|---|---|
| 6a | 1644, 2922, 1289, 3316, | 2.58 (3H, s), 3.61 (4H, s), 3.85 (4H, s), | 520 |
| 6.94–8.32 (10H, m), 10.22 (1H, s) | |||
| 6b | 1635, 2957, 1261,1166, 3301 | 2.55 (3H, s), 3.61 (4H, d), 3.72 (4H, d), 3.88 (3H, s), | 471 |
| 6.93–8.80 (10H, m), 10.21 (1H, s) | |||
| 6c | 1634, 2934, 1298, 3287 | 2.22 (3H, s), 2.40 (4H, t) 2.57 (3H, s), 3.64 (4H, s), | 533 |
| 6.93–8.77 (10H, m), 10.20 (1H, s) | |||
| 6d | 1634, 2934, 1298, 1168, 3287 | 2.39 (3H, s), 2.55 (3H, s), 2.64 (4H, s), 3.71(4H, d), | 484 |
| 3.89 (3H, s), 6.96–8.79 (10H, m), 10.21 (1H, s) | |||
| 6e | 1644, 2922, 1289, 3316 | 2.50 (4H, s), 2.57 (3H, s), 3.53 (2H, s), 3.65 (4H, s) | 609 |
| 6.92–8.76 (15H, m), 10.20 (1H, s) | |||
| 6f | 1644, 2922, 1289, 1168, 3316 | 2.45 (4H, t), 2.55 (3H, s), 3.53 (2H, s), 3.64 (4H, t), | 560 |
| 3.88 (3H, s), 6.91–8.76 (15H, m), 10.18 (1H, s) | |||
| 6g | 1635, 2914, 1261, 3301 | 2.57 (3H, s), 2.63 (4H, s), 4.03 (4H, s), | 536 |
| 7.66–8.32 (10H, m), 10.69 (1H, s). | |||
| 6h | 1635, 2914, 1261, 1166, 3301 | 2.56 (3H, s), 2.63 (4H, t), 3.87 (3H, s), 4.01 (4H, t), | 487 |
| 6.95–8.78 (10H, m), 10.20 (1H, s) | |||
| 6i | 1642, 2933, 1291, 3301 | 1.98 (4H, s), 2.57 (3H, s), 3.47 (4H, s), | 504 |
| 6.53–8.76 (10H, m), 10.13 (1H, s). | |||
| 6j | 1636, 2928, 1321, 3305 | 1.97 (4H, s), 2.55 (3H, s), 3.41(4H, s), 3.87 (3H, s), | 455 |
| 6.53–8.77 (10H, m), 10.13 (1H, s) | |||
| 6k | 1642, 2933, 1291, 3301 | 1.55 (4H, s), 1.63 (2H, m), 2.57 (3H, s), 3.47 (4H, s), | 518 |
| 7.02–8.76 (10H, m), 10.16 (1H, s). | |||
| 6l | 1637, 2932, 1289, 3312 | 1.55 (4H, s), 1.64 (2H, m), 2.55 (3H, s), 3.66 (4H, s), | 469 |
| 3.88 (3H, s), 7.10–8.76 (10H, m), 10.19 (1H, s) | |||
| 6m | 1635, 291, 1281, 3311 | 2.57 (3H, s), 2.93 (2H, s), 3.91 (2H, s), 4.81 (2H, s), | 566 |
| 7.21–8.82 (14H, m), 10.21 (1H, s). | |||
| 6n | 1636, 2933, 1291, 3320 | 2.55 (3H, s), 2.93 (2H, t), 3.84 (2H, t), 3.90 (3H, s), | 517 |
| 4.82 (2H, t), 7.12–8.81 (14H, m), 10.20(1H, s) | |||
| 6o | 1644, 2933, 1288, 3300 | 2.51 (3H, s), 3.18 (4H, s), 3.69 (4H, s), | 596 |
| 7.45–8.99 (14H, m), 10.70 (1H, s) | |||
| 6p | 1635, 2923, 1291, 3305 | 2.55 (3H, s), 3.28 (4H, s), 3.64 (2H, s), 3.77 (2H, s), | 547 |
| 3.84 (3H, s), 6.63 – 9.00 (14H, m), 10.68 (1H, s) | |||
| 6q | 1645, 2945, 1288, 3320 | 1.65 (4H, t), 2.57 (3H, s), 3.76 (4H, t), 3.93 (4H, t), | 576 |
| 6.98–8.78 (10H, m), 10.19 (1H, s). | |||
| 6r | 1645, 2930, 1290, 3306 | 2.44 (2H, t), 2.55 (3H, s), 3.38 (4H, t), 3.53 (2H, t), | 514 |
| 3.63 (4H, t), 3.88 (3H, s), 6.92–8.77 (10H, m), 10.18 (1H, s). |
Table 2: Spectral data of synthesized benzofuran derivatives
A series of 18 new compounds were synthesized. The structural data of all benzofuran derivatives is shown in Table 1. Scheme 1 illustrates the preparation of target compounds. The structure of the synthesized compounds was elucidated by IR, 1H NMR, mass spectral data and elemental analysis.

Scheme 1: Synthetic route for the preparation of novel benzofuran derivatives
General method of synthesis of N-(2-aroyl)-3-methyl-1-benzofuran-
5-yl)-6-aminoalkylnicotinamide 6(a-r)
In the 1H NMR spectra of the compounds, NH proton of the benzofuran ring was seen as singlet at about 10.20- 10.70 δ ppm. Signal due CH3 of benzofuran appeared at 2.55-2.58 δ ppm as a singlet. All other aromatic and aliphatic protons were observed in the expected regions. Mass spectra of all compounds showed M+ peaks in agreement with their molecular formula. In the IR spectra of all compounds C=O stretching bands were observed at 1635-1644 cm-1 and stretching band of CH3 of benzofuran ring observed at 2914-2932 cm-1.
The benzofuran derivatives were evaluated for their in vitro antimicrobial activity. MICs were recorded as the minimum concentration of compound, which inhibit the growth of tested microorganisms. Antimicrobial activities of the compounds were tested using Mueller-Hinton broth (Hi Media M 391) medium. Microbial strains used for testing included, Staphylococcus aureus (NCIM 5021) and Salmonella typhimurium (NCIM 2501). All test compounds were found to be inactive against above bacterial strains.
The in vitro antioxidant activity of the test compounds was determined by DPPH method using L-ascorbic acid (an antioxidant agent) as a positive control. The compounds were tested for antioxidant activity at 200, 100 and 50 μg/ml concentration. Amongst the compounds screened for antioxidant activity, 6a, 6b, 6d, 6h, 6o, 6p and 6r showed very good antioxidant activity as shown in Table 3.
| Concentration in µg/ml | 200 | 100 | 50 |
|---|---|---|---|
| L-ascorbic acid | 99.2 | 99 | 98.8 |
| 6a | 95.3 | 95.3 | 91.9 |
| 6b | 100 | 98.3 | 88.1 |
| 6d | 94.6 | 80.2 | 28.7 |
| 6g | 57.9 | 48.0 | 28.2 |
| 6h | 98.1 | 91 | 89.8 |
| 6o | 97.9 | 94 | 85.7 |
| 6p | 100 | 97.2 | 93.6 |
| 6r | 94.2 | 93.2 | 92.6 |
Table 3: % Antioxidant activity of the compounds
The compounds with morpholine, 1-pyridyl-2-yl piperazine at R1 and with Br and OMe at R showed very significant antioxidant activity. While the compounds with thiomorpholine and piperzin-1-yl ethanol at R1 and OMe at R also showed good antioxidant activity. Compounds with pyrrolidine, piperidine and N-benzylpiperazine at R1 do not show any antioxidant activity.
References
- Jinno S, Otsuka N. Total synthesis of a natural antioxidant and structure activity relationships of related compounds. Chem Pharm Bull 1999;47:1276-83.
- Masataka F, Nobuo M, Tsutomu H, Tomihiro N. Antioxidant activities of vitamin E derivative having benzofuranskelton. Nippon KagakkaiKoenYokoshu 2002;81:1381.
- Bindoli A, Rigobello MP, Musacchio E, S curi R, Rizzoli V, Galzigna L. Protective action of a new benzofuran derivative on lipid peroxidation and sulphydrylgroups oxidation. Pharmacol Res 1991;24:369-75.
- Lu Z, Gregory R. Potent, selective, orally bioavailable inhibitors of tumor necrosis factor-α converting enzyme (TACE): Discovery of indole, benzofuran, imidazopyridine and pyrazolopyridine P1′ substituents. Bioorg Med ChemLett 2008;18:1958-62.
- Kirilmis C, Ahmedzade M, Servi S. Synthesis and antimicrobial activity of some novel derivatives of benzofuran: Part 2, The synthesis and antimicrobial activity of some novel 1-(1-benzofuran-2-yl)-2-mesitylethanone derivatives. Eur J Med Chem 2008;43:300-8.
- Rizzo S, Rivière C, Piazzi L, Bisi A, Gobbi S, Bartolini M. Benzofuran-based hybrid compounds for the inhibition of cholinesterase activity, beta amyloid aggregation, and abeta neurotoxicity. J Med Chem 2008;51:2883-6.





