- *Corresponding Author:
- R. Sahu
Department of Pharmacy, School of Medical & Allied Sciences, Galgotias University, Greater Noida 201310, India
E-mail: rakeshsahu1100@gmail.com
| Date of Received | 15 November 2020 |
| Date of Revision | 14 February 2021 |
| Date of Acceptance | 15 March 2021 |
| Indian J Pharm Sci 2021;83(2):162-185 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
Pyridine is an imperative pharmacophore, a privileged scaffold and an exceptional heterocyclic system in the field of drug discovery which provides many opportunities in study/explore this moiety as an anticancer agent by acting on various receptors of utmost importance. Several pyridine derivatives are reported to inhibit tubulin polymerization, androgen receptors, human carbonic anhydrase, kinase, topoisomerase enzyme and many other targets for controlling and curing global health issue of cancer. Now a days in combination with other moieties researchers are focusing for development of pyridine new entities for the treatment of cancer. This review throws light on recent biological expansions of pyridine along with their structure activity relationships/molecular docking to deliver association between various synthesized newer derivatives and receptor sites.
https://1xbetx.fun https://bahiscom.fun https://casibomagiris.com https://cratosslot.fun https://grandpashabet.fun https://holiganbet.fun https://holiganbet-giris.info https://jojobet.fun https://jojobetgiris.xyz https://kralbet.fun https://meritking.fun https://vdcasino.fun https://onwin.direct https://cypocafe.com https://sahabetgiris.fun
Keywords
Pyridine moiety, anticancer, heterocyclic compound, cancer treatment
Cancer is a global health issue that affects a vast portion of the human population and is regarded as the uncontrolled growth of cells in the body, which is one of the most challenging and complex diseases to treat[1,2] . It is considered as another major cause of the death after cardio vascular disorders worldwide[3]. In 2018, world health organization (WHO) reported that 18.1 million people around the globe had cancer and 9.6 million deceased from the disease. By 2040, these records will nearly twice, with the highest increase in low and middle income countries, where greater than two thirds of the world’s tumors will occur[4]. Cancer is the cause of almost 30 % of all hasty deaths from non-communicable diseases among adults aged 30 to 69. Among all, the most frequently diagnosed cancer is lung (11.6 % of all cases), followed by female breast (11.6 %) and colorectal cancers (10.2 %). Lung cancer is the foremost cause of death from cancer (18.4 % of all deaths), followed by colorectal (9.2 %) and stomach cancers (8.2 %). The most usual cause of cancer is tobacco usage, which accounts for 25 % of all cancer deaths around globally. Hence cancer is a serious issue hampering human health. So, as to tackle them new various anticancer agents are being developed ever now and then. Still their toxicity profile has restricted their clinical use as anticancer agents[5]. Therefore, further exploration of new chemo therapeutic agents with great efficacy and least adverse effects is critically essential for medicinal chemists.
Anticancer drugs are categorized into several groups based on their mechanism of action that is biosynthetic blocker of nucleic acid, transcription interferer, structural and functional of Deoxyribonucleic acid (DNA) interferer, interferer of protein synthesis and function, influencer of hormone homeostasis; or modulator of immune system[6]. To decrease toxic effects of antitumor agents towards normal cells, specific targeted drug therapy is chosen which is considered to hit tumor cells only[7]. As specified by the USA national cancer institute drug repository, 227 endorsed antitumor treatments are at present avowed to treat roughly 40 types of malignancies[6,8]. Because of great demand of antitumor medications (tons/y) investigators are going on in the way to develop new and effective treatment for tumor[8]. For this reason some researchers or scholars looked into the effect produced by various compounds containing pyridine moiety.
Pyridine compounds (fig. 1) are well defined by the presence of a six membered heterocyclic ring with the chemical formula C5H5N, comprising of five carbon atoms and one nitrogen atom. In many aspects it can be correlated to well recognized and fundamental aromatic benzene molecule, with one C-H group changed by a nitrogen atom. It was first isolated from bone oil and coal tar and characterized by Anderson in 1846. The cyclic nature of pyridine was identified by Dewar and Korner in 1869[9]. It plays an important role by catalyzing both biological and chemical systems[10]. It has a conjugated arrangement of six π electrons precisely as benzene has, that are delocalized about the heterocyclic ring. The molecule is planar in nature and follows Huckel standards for aromaticity[11,12]. Additionally, pyridines are a class of both synthetically and naturally occurring heterocyclic compounds and its nucleus is a well-studied six membered heterocyclic moiety with a wide range of biological and therapeutic applications[13-15]. Moreover, the current interest in the development of new anticancer agents can be partially ascribed to the increasing appearance of multidrug resistance and adverse side effects are a serious threat to public health[16-18]. Therefore, the development of new and efficacious medications is a very significant goal and most of the researcher’s efforts in this field are directed towards the design of new agents. It is reported that some important anticancer drugs possess a pyridine nucleus. Thus, this study gives promising compounds possessing a pyridine nucleus that can be investigated for future in vivo and clinically oriented studies[19-21]. Some of the physical properties and marketed products of pyridine containing antitumor drugs are mentioned in Table 1[22] and Table 2[23], whereas in Table 3[24-50] patents on pyridine are mentioned respectively.

Figure 1: Chemical structure of pyridine
| Formula | Molecular weight | Density | Melting point | Boiling point | Refractive | Index solubility |
|---|---|---|---|---|---|---|
| C5H5N | 79.102 Da | 0.9819 g/ml | −41.6º | 115.2º | 1.5093 | Miscible in all proportions with water and most common organic solvents |
Table 1: A Physical Property of The Pyridine [22]
| S. No. | Drug | Brand and company | Primary target | Food and drug administration (FDA) approval | Structure |
|---|---|---|---|---|---|
| 1. | Apalutamide | ERLEADA, Janssen Biotech Inc. | Androgen receptor inhibitor | In September, 2019 | 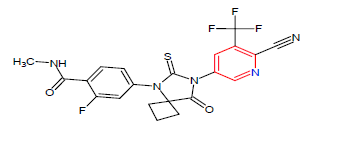 |
| 2. | Pexidartinib | TURALIO, Daiichi Sankyo | Kinase inhibitor | In August, 2019 | 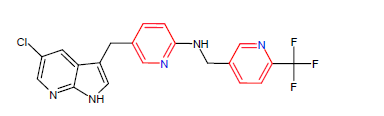 |
| 3. | Alpelisib | PIQRAY, Novartis Pharmaceuticals Corporation | Kinase inhibitor | In May, 2019 | 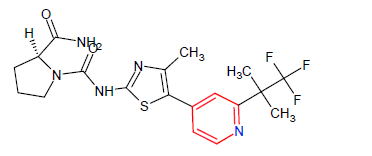 |
| 4. | Venetoclax | VENCLEXTA, AbbVie Inc. and Genentech Inc. | BCL-2 inhibitor | In May, 2019 | 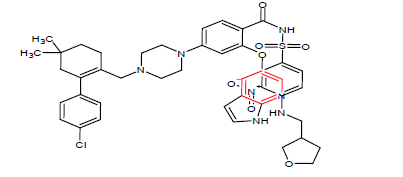 |
| 5. | Ivosidenib | TIBSOVO, Agios Pharmaceuticals Inc. | IDH1 inhibitor | In May, 2019 | 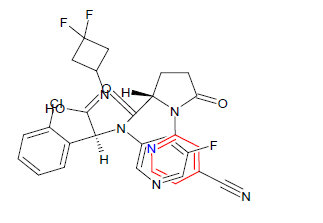 |
| 6. | Lorlatinib | LORBRENA, Pfizer Inc. | Kinase inhibitor | In November, 2018 | 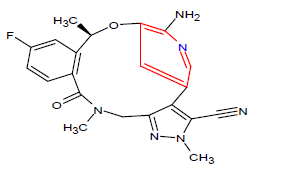 |
| 7. | Netarsudil | RHOPRESSA, Aerie Pharmaceuticals | Rho kinase inhibitor |
In December, 2017 | 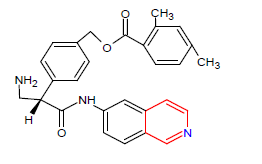 |
| 8. | Acalabrutinib | CALQUENCE, AstraZeneca | Kinase inhibitor | In October, 2017 | 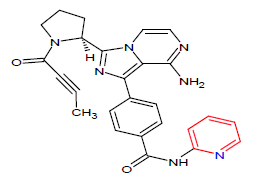 |
| 9. | Abemaciclib | VERZENIO, Eli Lilly and Company | Kinase inhibitor | In September, 2017 | 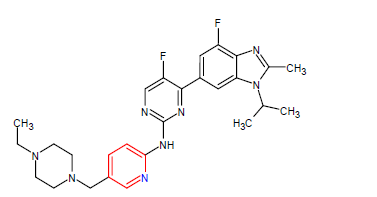 |
| 10. | Enasidenib | IDHIFA, Celgene Corp. | IDH2 inhibitor | In August, 2017 | 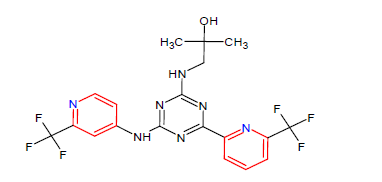 |
| 11. | Neratinib | NERLYNX, Puma Biotechnology Inc. | Kinase inhibitor | In July, 2017. | 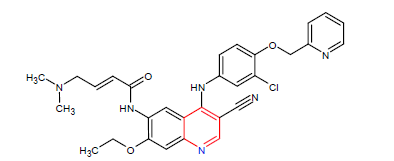 |
Table 2: Approval of More Recently Marketed Drugs Containing Pyridine [23]
| S.No. | Patent no. | Patent date | Inventors | Description |
|---|---|---|---|---|
| 1. | WO2020039097A1 [24] | February 27, 2020 | Zoe Cournia, Argiris Efstratiadis, Anna Kapella, Elias Couladouros, Savvas Christoforidis | 2,6-bis(((1h-benzo[d]imidazol-2-yl)thio)methyl)pyridine and n2,n6-dibenzylpyridine-2,6-dicarboxamide derivatives and related compounds as PI3K inhibitors for treating cancer |
| 2. | US20200062754A1 [25] | February 27, 2020 | Xiong Cai, Changgeng Qian, Haixiao Zhai | Fused amino pyridine as hsp90 inhibitors |
| 3. | WO2020039060A1 [26] | February 27, 2020 | Carsten Schultz-Fademrecht, Bert Klebl, Peter Nussbaumer, Carsten Degenhart, Matthias Baumann | 4-substituted pyrrolo[2,3-b]pyridine as erbb modulators useful for treating cancer |
| 4. | WO2020033288A1 [27] | February 13, 2020 | Michelle Machacek, David Witter, Craig Gibeau, Chunhui Huang, Shuhei Kawamura, David L. Sloman, Phieng Siliphaivanh, Ryan Quiroz, Murray Wan, Sebastian Schneider, Charles S. Yeung, Michael H. Reutershan, Timothy J. Henderson, Jean-Laurent Paparin, Houcine Rahali, Jonathan M. E. Hughes, Sulagna Sanyal, Yingchun Ye, David A. Candito, Patrick S. Fier, Steven M. Silverman | PRMT5 inhibitors |
| 5. | WO2020030925A1 [28] | February 13, 2020 | Peter Blencowe, Mark Charles, Andrew Cridland, Tennyson Ekwuru, Robert Heald, Ellen Macdonald, Hollie Mccarron, Laurent Rigoreau | Heterocyclic substituted ureas, for use against cancer |
| 6. | US20200040002A1 [29] | February 06, 2020 | Yun-Long Li, Wenyu Zhu, Song Mei, Joseph Glenn | Tricyclic fused thiophene derivatives as JAK inhibitors |
| 7. | US20200022983A1 [30] | January 23, 2020 | Jay Copeland Strum, John E. Bisi, Patrick Joseph Roberts, Jessica A. Sorrentino | Treatment of RB-negative tumors using topoisomerase inhibitors in combination with cyclin dependent kinase 4/6 inhibitors |
| 8. | CN110698491A [31] | January 17, 2020 | Zhang Sanqi, Fan Ye, Cao Yongxiao, Shen Ying, Yang Xueyan, Xin Minxing, Cao Lei | 2- (camptothecin-10-oxyl) acetamide compound and application thereof |
| 9. | WO2020014465A1 [32] | January 16,2020 | Alfredo C. Castro, David T. Jonaitis | Polymorphic compounds and uses thereof |
| 10. | US20200010458A1 [33] | January 09,2020 | Noriyasu Haginoya, Takashi Suzuki, Miho Hayakawa, Masahiro Ota, Tomoharu Tsukada, Katsuhiro Kobayashi, Yosuke Ando, Takeshi Jimbo, Koichi Nakamura | Pyridone derivatives having tetrahydropyranylmethyl groups |
| 11. | US20200010420A1 [34] | January 09, 2020 | Janet L. Gunzner-Toste, Daniel Sutherlin, Mark S. Stanley, Liang Bao, Georgette M. Castanedo, Rebecca L. Lalonde, Shumei Wang, Mark E. Reynolds, Scott J. Savage, Kimberly Malesky, Michael S. Dina, Michael F.T. Koehler | Pyridyl inhibitors of hedgehog signalling |
| 12. | US20200002310A1 [35] | January 02, 2020 | Michael Joseph Luzzio, Kevin Daniel Freeman-Cook, Samit Kumar Bhattacharya, Matthew Merrill Hayward, Catherine Angela Hulford, Christopher Lowell Autry, Xumiao Zhao, Jun Xiao, Kendra Louise Nelson | Sulfonyl amide derivatives for the treatment of abnormal cell growth |
| 13. | US20190367456A1 [36] | December 05, 2019 | David G. Hangauer, Jr. | Biaryl compositions and methods for modulating a kinase cascade |
| 14. | US20190367507A1 [37] | December 05, 2019 | Prabha N. Ibrahim, Wayne Spevak, Jiazhong Zhang, Songyuan Shi, Ben Powell, Yan Ma | Heterocyclic compounds and uses thereof |
| 15. | US20190352279A1 [38] | November 21, 2019 | Sunil Kumar KC, John Hood | 3-(benzoimidazol-2-yl)-indazole inhibitors of the Wnt signaling pathway and therapeutic uses thereof |
| 16. | US20190343814A1 [39] | November 14, 2019 | Alexander Sokolsky, Oleg Vechorkin, Kai Liu, Jun Pan, Wenqing Yao, Qinda Ye | Pyrazolopyridine compounds and uses thereof |
| 17. | US20190336506A1 [40] | November 07, 2019 | Adnan M. M. Mjalli, Bapu Gaddam, Dharma Rao Polisetti, Matthew J. Kostura, Mustafa Guzel | Tricyclic compounds as modulators of TNF-α synthesis and as PDE4 inhibitors |
| 18. | US20190135785A1 [41] | May 09, 2019 | Takayuki Inukai, Jun Takeuchi, Tomoko Yasuhiro | Quinoline derivative |
| 19. | EP2393808A1 [42] | May 08, 2019 | Douglas Phillipson, Katharina Reichenbacher, Robert J. Duguid, Jacqueline A. Ware | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| 20. | US20190106439A1 [43] | April 11, 2019 | Eric Jon Jacobsen, James Robert Blinn, John Robert Springer, Susan Landis Hockerman | Heterocyclic itk inhibitors for treating inflammation and cancer |
| 21. | RU2682245C1 [44] | March 18, 2019 | John Hood, David Mark Wallace, Sunil Kumar Ks | Indazolie Wnt signal pathway inhibitors and their therapeutic applications |
| 22. | US20180235948A1 [45] | August 23, 2018 | Erkan Baloglu, Sharon Shacham, William Senapedis | (s,e)-3-(6-aminopyridin-3-yl)-n-((5-(4-(3-fluoro-3-methylpyrrolidine-1-carbonyl)phenyl)-7-(4-fluorophenyl)benzofuran-2-yl)methyl)acrylamide for the treatment of cancer |
| 23. | US20180228831A1 [46] | August 16, 2018 | Alyssa M. Larson, Kevin Love, Alisha K. Weight, Alan Crane, Robert S. Langer, Alexander M. Klibanov | Polysaccharide and nucleic acid formulations containing viscosity-lowering agents |
| 24. | US20180228782A1 [47] | August 16, 2018 | Sunil Kumar KC, David Mark Wallace, Jianguo Cao, Chandramouli Chiruta, John Hood | 3-(1H-pyrrolo[2,3-B]pyridin-2-yl)-1H-pyrazolo[3,4-C]pyridines and therapeutic uses thereof |
| 25. | US20170260198A1 [48] | September 14, 2017 | Marion Hitchcock, Anne Mengel, Vera Pütter, Gerhard Siemeister, Antje Margret Wengner, Hans Briem, Knut Eis, Volker Schulze, Amaury Ernesto Fernandez-Montalvan, Stefan Prechtl, Simon Holton, Jorg Fanghanel, Philip Lienau, Cornelia Preusse, Mark Jean Gnoth | Substituted benzylindazoles for use as BUB1 kinase inhibitors in the treatment of hyperproliferative diseases |
| 26. | US20170174653A1 [49] | June 22, 2017 | Brian A. Sherer, Nadia Brugger | TLR7/8 antagonists and uses thereof |
| 27. | AU2015299173A1 [50] | February 09, 2017 | Benjamin Bader, Wilhelm Bone, Hans Briem, Uwe Eberspacher, Knut Eis, Joanna Grudzinska-Goebel, Marcus Koppitz, Julien Lefranc, Philip Lienau, Ulrich Lucking, Dieter Moosmayer, Hans Schick, Gerhard Siemeister, Franz Von Nussbaum, Antje Margret Wengner, Lars Wortmann | 2-(morpholin-4-yl)-l,7-naphthyridines |
Anticancer profile of pyridine Based derivatives
Recently reported some synthetic new compounds that have a promising and potential activity against cancer cells and has pyridine moiety as essential part in those structure are mentioned here along with their structure activity relationship (SAR) or molecular docking or both studies and acting targets or inhibitory target.
Migration and Ras related C3 botulinum toxin substrate 1 (RAC1) inhibitors:
A cancer cell finishes a series of stages including migration from the primary cancer, surrounding tissues invasion and intravasation through basement membranes, survival during circulation and arrest at a distant target organ[51]. The Ras-related C3 botulinum toxin substrate 1 (RAC1) gene activates multiple signaling pathways that lead to uncontrolled proliferation, is a member of the Ras-related C3 botulinum toxin substrate (RAC) subfamily of guanosine triphosphate enzyme (GTPase). It has been involved in the regulation of cellular migration and invasion in breast tumor cells. Rac1 is activated by Guanine nucleotide exchange factors (GEFs) that are regulated by many of cell surface receptors. Hence, therapeutic approaches that inhibit binding of GEFs to Rac1 are a rational means to migration inhibition of cancer cells[52-55].
Vlaar et al.[56] explored a carbozole derivatives in order to develop new anti-migratory agents to prevent the malignance cell spreading by inhibiting migration and RAC1. Compounds 1a and 1b (fig. 2) inhibit migration activity of metastatic cell line MDA-MB-231 (Breast carcinoma cell line) by 32 % and 34 %, respectively. Furthermore compound 1b was shown to inhibit activation of the Ras homologous (Rho) GTPase Rac1 by 55 % at 0.25 μM in both MDA-MB-231 and MDAMB- 435 (Breast carcinoma cell line) cell lines.

Figure 2: Docked structure and SAR of carbozole derivatives
Indoleamine 2,3-dioxygenase 1 inhibitors:
The metabolism of Tryptophan (Trp) occurs through two independent pathways: the kynurenine pathway (KP) that comprises of the oxidative ring opening of the pyrrole moiety of the indole as the initiating step and the melatonin pathway that first introduces a hydroxyl group to the indole followed by subsequent biochemical alterations of the hydroxyl and the carboxyl functional groups to produce the hormone melatonin and the neurotransmitter serotonin. The rate limiting step of the KP is catalyzed by the heme-containing Indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase, the product N-formyl kynurenine from which is rapidly converted to kynurenine. In mammalians, there are two known isoforms of IDO as IDO1 and IDO2. Among them, IDO2 is the least recognized and its functional role is yet to be fully defined. IDO1 is well defined and recognizes a number of indole type compounds like L/D-Trp and melatonin. IDO1 is up regulated in immune cells by the proinflammatory cytokine Interferon-γ (IFN-γ), which works as a mechanism to prevent overactive immunological responses, hence it prevents damages to host’s tissues and organs[57-60].
Kong et al.[61] designed and synthesized a series of 1H indole-4,7-dione derivatives and evaluated their inhibitory activity in IDO1 and in IFN-γ stimulated Hela (cervical cancer cell line) cells. The SAR and enzyme kinetics experiments revealed that N alkyl substituted side chain of 5-(pyridin-3-yl)-1H-indole- 4,7-dione (compound 2) has most promising inhibitory activity (50 % inhibitory concentration value or half maximal inhibitory concentration (IC50) of 0.16 ±0.02 μM in enzymatic assay) against IDO1 enzyme (fig. 3). Moreover, it has been indicated that this type of IDO1 inhibitors might be a kind of reversible competitive inhibitors.

Figure 3: Docked structure and SAR of 1H-indole-4,7-dione (2) derivative
Yang et al.[62] synthesized and evaluated two series of naphthoquinone derivatives, naphthoindolizine and indolizinoquinoline-5,12-dione derivatives for their IDO1 inhibitory activity. Many of the compounds showed significant inhibition potency and high selectivity for IDO1 over tryptophan 2,3-dioxygenase and the SAR was also summarized. Among all, the compound 3a (fig. 4) showed promising and most potent with IC50 value 0.23 μM against IDO1 enzyme. However, compound 3b was also identified as promising lead compound with IC50 value 0.372 μM against HeLa cell. Furthermore, molecular docking study revealed that it has good interaction with protein and can be useful for further design of new IDO1 inhibitors as antitumor agents.

Figure 4: Docked structure and SAR of naphthoindolizine (3a) and indolizinoquinoline (3b) 5,12-dione derivatives
Human carbonic anhydrase IX/XII inhibitors:
Carbonic anhydrase (CAs) is an important target for cancer treatment because of its limited aspects in the normal tissues and predominant expression in varieties of cancer cells[63]. CAs is omnipresent metallo enzymes which catalyze the reversible hydration of carbon dioxide into protons and bicarbonate. Among the seven CA families, αCA are only enzyme subfamily found in human and further categorized in 15 varied human isoforms playing critical roles in a multitude of physiological functions and pathological processes for example respiration and transport of CO2 and bicarbonate between metabolizing tissues and lungs, homeostasis of pH and CO2, secretion of electrolytes in various tissues and organs, biosynthetic reactions (i.e., gluconeogenesis, lipogenesis and ureagenesis), bone resorption, calcification and tumor growth[64,65]. Human CA I and II are cytosolic proteins with high enzymatic efficiency that are constitutively expressed in all tissues. Conversely, human CA IX and XII are membrane related proteins that can be over expressed in the hypoxic cancer environment, with CA IX not considerably present in the majority of healthy tissues[64,66]. Furthermore, it contributes to tumor progression by stimulating tumor cell migration, adhesion and invasion[67]. Now a days CAs has been recognized as a significant biomarker of hypoxia and its over expression is frequently linked with a poor responsiveness to the classical radio and chemotherapies[68].
Uslu et al.[69] prepared a series of 2-arylbenzimidazole derivatives containing sulfonamide functionality as well as carboxylic acid, hydroxamic acid, carboxamide and boronic acid functionalities, which act as human CA inhibitors. The sulfonamide containing benzimidazole compound 4 derivatives (fig. 5) revealed intriguing inhibitory activity against cancer associated CA IX and XII with inhibitory constant (Ki) values in the range of 0.0052-0.0293 μM and 0.0099 -0.0417 μM, respectively. Notably, compound 4 was the most potent and selective CA IX (Ki=0.0066 μM) and XII (Ki=0.0099 μM) inhibitor with an important selectivity ratio over cytosolic CA I and II isoforms in the range of 0.0034-0.0252 μM.

Figure 5: Structure of 5-sulfonamide benzimidazole (4) with SAR
Ansari et al.[70] design, synthesis and evaluate a series of pyridine-thiazolidinone derivatives in order to obtain human CA IX inhibitors. The binding affinity of the compounds was measured using fluorescence binding studies and enzyme inhibition activity by esterase assay of CA IX. It was observed that compound 5a and 5b (fig. 6) significantly inhibit the CA IX activity with the IC50 values 1.61 μM and 1.84 μM, the binding affinity for CA IX was significantly great with their equilibrium dissociation constant (KD) values 11.21 μM and 2.32 μM, respectively. All the compounds were further screened in vitro for anticancer activity and found that compound 5a and 5b exhibit substantial antitumor activity against HepG-2 (human liver cancer cell line) and MCF-7 (Breast carcinoma cell line) cell lines.

Figure 6: Docked structure and SAR of pyridine-thiazolidinone derivatives
Mutant isocitrate dehydrogenase (IDH) inhibitors:
Isocitrate dehydrogenase (IDH) enzyme family, includes IDH1, IDH2 and IDH3, are key metabolic enzymes which can convert isocitrate to α-ketoglutarate (α-KG)[71,72]. Recently IDH mutations have been recognized in somatic tumor associated numerous hematologic and solid tumors, such as glioma, acute myeloid leukemia (AML), cholangiocarcinoma cancer, malignant chondrosarcomas 4 and others[73-75]. IDH mutations frequently occur at R132 in IDH1 or R140 and R172 in IDH2, which are situated in the catalytic pocket of these enzymes. Considerably, these mutations let IDH enzymes to gain an exceptional activity, which catalytically alters α-KG to an onco-metabolite 2-hydroxyglutarate. This onco-metabolite is intensely connected to impaired hematopoietic differentiation and stimulates leukemia due to its ability of overall DNA hyper methylation[76]. Pharmacological barrier of mutant IDH1 enzyme efficiently inhibits colony formation of IDH mutated patient derived AML cells but not that of normal CD34+ (cluster of differentiation 34 which is a glycosylated transmembrane protein) bone marrow cells[77]. By itself, mutant IDH has now become a therapeutic specified target of high interest in the field of anticancer drug discovery.
Liu et al.[78] synthesized and evaluated a series of 3-aryl- 4-indolylmaleimides in order to get IDH1/R132H inhibitors. New structure was acquired through highthroughput screening and structure-based optimization and most compounds displayed great inhibitory effects and were highly selective against IDH1/R132H. Biological evaluation of the activities and function at cellular level revealed that compounds 6a, 6b and 6c (fig. 7) were the most potent with IC50 values of 0.05 μM, 0.06 μM and 0.06 μM, respectively against IDH1/R132H in U87MG (Glioblastoma cell line) cells and great selectivity against IDH1/WT (wild type) and IDH2/WT. Furthermore, molecular modeling study provided information that can be useful for the design of new IDH1/R132H inhibitors as anticancer agents.

Figure 7: Docked structure and SAR of 3-aryl-4-indolylmaleimide derivative
Hu et al.[79] designed, synthesized and biologically evaluated a series of 3-(7-azainodyl)-4- indolylmaleimides for their IDH1/R132H inhibitory activities. Many compounds shown favorable inhibitory effects and were highly selective against the IDH1/ R132H. Evaluation of the biological activities at the cellular level showed that compounds 7a (IC50=0.40 μM), 7b (IC50=0.36 μM), 7c (IC50=0.16 μM), 7d (IC50=0.28 μM) and 7e (IC50=0.34 μM) effectively reduce the production of 2-hydroxyglutaric acid in U87MG cells in a dose independent manner, which associates well with their inhibitory activity toward IDH1/R132H. Preliminary SAR and molecular modeling studies (fig. 8) provided that these findings may offer new insights into the development of new IDH1/R132H inhibitors.

Figure 8: Docked structure and SAR of 3-(7-azainodyl)-4-indolylmaleimide derivative
Androgen receptor (AR) inhibitor:
The Androgen receptor (AR) belongs to the subfamily of steroid receptors, which includes the glucocorticoid and progesterone receptors. They all belong to the nuclear hormone receptor superfamily of ligand activated transcription factors. The AR comprises of a carboxyl (C-) terminal ligand binding domain (LBD), a central DNA binding domain (DBD) and the amino N terminal domain (NTD) protecting the major transactivation function[80-82] and it possesses an extremely conserved DBD, a moderately conserved LBD and a slight conserved NTD[83,84]. All three these domains are important for receptor functions. The biological functions of AR are initiated by binding with 5-dihydrotestosterone/testosterone, which leads to the AR conformational change and its translocation from the cytosol to the nucleus to stimulate transcriptional regulation of specific genes that further modulated by various AR coregulators[85,86]. The AR continuously play a stimulator role in several different prostate cancer cell types during progression of prostate cancer (PCa) and metastasis[87]. In the beginning of the treatment, the insufficiency of androgens leads to cancer regression. However, eventually during hormone therapy, PCa develops into a castration resistant stage but remains AR dependent[88,89]. Inactivation of the AR also in castration resistant PCa seems to be therefore a key target of treatment. Current therapeutics include steroidal and non-steroidal anti androgens targeting typically LBD of AR[90-92]. Although other modulators targeting another domain are speedily investigated and shows beneficial preclinical profiles[84,93].
Shi et al.[92] developed and synthesized a series of steroidal pyridines derivative through the base promoted three component reaction and preliminarily evaluated for their anti-proliferative activity against cancer cell lines. SARs studies displayed that the 4th position of the pyridine ring were chosen over the phenyl rings for the activity. Among these compounds, 8 (fig. 9) possessing an extra pyridine ring at the p position demonstrated good growth inhibition against cancer cells, especially for PC-3 (prostate cancer cell line) cells with an IC50 value of 1.55 μM. For the AR+ sensitive PCa cell line LNCaP (prostate cancer cell line), compound 8 inhibited growth with an IC50 value of 8.48 μM, slightly less potent than the anti-prostate cancer drug abiraterone (IC50=3.29 μM). Further mechanistic studies showed that it inhibited colony formation, migration and evasion of PC-3 cells in a concentration dependent manner as well induced apoptosis of PC-3 cells possibly via the mitochondria associated apoptotic pathways.

Figure 9: Structure of steroidal pyridine derivative with SAR
Aldo keto reductase (AKR) inhibitors:
The Aldo keto reductase (AKRs) is a distinct superfamily of proteins that catalyze the reduction of carbonyl groups as well steroid double bonds in the presence of NADPH[94]. It comprises various genes, among them AKR1C1-AKR1C4 genes are situated on chromosome 10 p15-p14 and include of 12 exons. The AKR1C4 and AKR1C3 are almost exclusively involve in the liver and PCa, AKR1C1 and AKR1C2 are most distinguished in the mammary glands includes breast cancer, endometrial cancer, colorectal cancer.
The AKR1C3 (also known as prostaglandin-F synthase) protein catalyzes the conversion of prostaglandins (PGF) H2 and D2 into Prostaglandin F2 alpha (PGF2α) and 9α, 11β-PGF2α respectively. It has the highest catalytic effectiveness than the AKR1C enzymes to interconvert testosterone with 14-androstene 3,17-dione. AKR1C2, is also known as bile acid binding protein and preferentially reduces Dihydrotestosterone (DHT) to the weak metabolite 5α-androstane-3α,17β- diol (3α-diol) without conversion of 3α-diol to DHT in the PC-3 cell line[95-97].
Savic et al.[98] synthesized and validated new A ring pyridine fused androstanes in 17a-homo-17-oxa (D-homo lactone), 17α-picolyl or 17(E)-picolinylidene series for potential antitumor activity in vitro using human tumor cell lines and recombinant targets of steroidal antitumor drugs. SAR revealed that pyridine fusion to position 3, 4 of the A ring intensely enhance affinity of 17α-picolyl compounds for cytochrome (CYP) 17 while conferring selective anti-proliferative activity against PC-3 cells. Similarly, pyridine fusion to the A ring of steroidal D-homo lactones led to recognition of new inhibitors of AKR1C3. Among them, compound 9 (fig. 10) found to be most promising with IC50 9.13 and 11.77 μM in MDA-MB-231 and HeLa cell respectively. Additionally molecular docking also suggested that compound 9 has higher binding affinity toward AKR1C3 (9.8 kcal/mol) similar to androstenedione.

Figure 10: Docked structure and SAR of pyridine fused 17α-picolyl derivative
Kinase inhibitor:
Kinase inhibitors have played an increasingly key role in the treatment of tumor and other diseases. Currently, more than 25 oncology medications that target kinases have been approved and many drugs therapeutics are in various stages of clinical evaluation[99]. Kinases are enzymes that transfer a phosphate group to a protein whereas phosphatases remove a phosphate group from protein[100]. Almost 538 known kinases are encoded in the human genome and these kinases uphold cellular function by turning protein function on, whereas corresponding phosphatases reverse this action[101,102]. Recent advances demonstrated that molecular mechanisms underlying cancer cell signaling have elucidated a critical role for kinases in the carcinogenesis and metastases of various types of cancer[103]. Since most protein kinases promote cell survival, proliferation and migration, when constitutively overexpressed or active, they are also linked with oncogenesis[104]. Over the past decades, multiple human malignancies have been identified to be associated with modulation and dysfunction of protein and lipid kinases and deactivated phosphatases on account of chromosomal reshuffling and genetic mutations[105,106]. Thus, some example of kinase inhibitors mentioned below that have been recently developed or reported as potential inhibitors of kinase.
Nawaz et al.[107] designed, synthesized and evaluated pyrazoline linked carboxamide derivatives for potential epidermal growth factor receptor (EGFR) kinase inhibitors, apoptotic, cardiomyopathy toxicity and other antitumor activity. Among all the synthesized compounds, 10a and 10b shown (fig. 11) good broad spectrum in vitro anticancer activity arrayed with 3-pyridine and the 4-pyridine group respectively, against the A549 (Lung adenocarcinoma cell line) (IC50 10.3±1.07 and 4.6±0.57 μM) and HCT-116 (Colon cancer cell line) (IC50 12.9±1.76 and 6.5±0.60 μM) cancer cell lines by using doxorubicin as the standard drug. Additionally, compounds 10a and 10b were exhibited to induce apoptosis in A549 cancer cells as evidenced by 4′,6-diamidino-2-phenylindole staining and apoptosis assay, which further strengthened in vitro anticancer findings.

Figure 11: Docked structure and SAR of pyrazoline-linked carboxamide derivatives
Yu et al.[108] designed, synthesized and biologically evaluated a series of imidazo[1,2-a]pyridine derivatives for potential phosphoinositide 3-kinase (PI3K) and mammalian target of rapamycin (mTOR) dual inhibitors as an effective targeting pathway for tumor therapy. Among the synthesized compounds, compound 11 was proved to be a potent PI3K/mTOR dual inhibitor with exceptional kinase selectivity, modest plasma clearance and acceptable oral bioavailability, with IC50 0.01, 0.052, 0.04, 0.071, 0.0052, 15.53 μM against various HCT-116, HT-29 (Colon cancer cell line), MCF-7, PC- 3, LOVO (Colon cancer cell line) and HUVEC (Human umbilical vein endothelial cells) cell lines respectively. Besides, compound 11 displayed significant inhibition with IC50 0.20 0.58 1.20 0.50 21 nM against PI3Kα, PI3Kβ, PI3Kγ, PI3Kδ and mTOR respectively. The docking results also revealed the inhibitory activity of compound 11 via forming various interactive bonds with PI3Kα and mTOR (fig. 12).

Figure 12: Docked structure and SAR of imidazo[1,2-a]pyridine derivative
Ullah et al.[109] synthesized and evaluated a series of pyridine-pyrazole-benzenethiourea and pyridinepyrazole- benzenesulfonamide scaffold for their inhibitory effect on human nucleotide Ectonucleotide pyrophosphatase/phosphodiesterase (ENPP) 1 and ENPP3 isoenzymes. Among them, compound 12a was the most potent inhibitor of ENPP3 (IC50=0.21 μM) and compound 12b was much selective to ENPP1 with IC50 value of 0.40 μM and found promising cytotoxic against HeLa, MCF-7 and 1321N1 (Astrocytoma cell line) cell lines. However, 12b displayed preferential cytotoxicity against MCF-7 (IC50=16.05 μM), which is similar to the potency of cisplatin. Additionally, docking results showed considerable binding interactions with active sites of ENPP isoenzymes (fig. 13).

Figure 13: Docked structure and SAR of pyridine-pyrazole-benzenethiourea (12a) and pyridine-pyrazole-benzene sulfonamide (12b) derivatives
B-cell lymphoma 2 (Bcl-2) inhibitors:
Apoptosis is a form of planned cell death that is activated in response to developmental cues or cellular stress. This selective cell suicide shows a crucial role in various physiological and pathological processes including growth, immunity and disease by the elimination of damaged or unnecessary cells helps to ensure organs health[110]. B-cell lymphoma 2 (Bcl-2) is a family of proteins responsible as key regulators of cell death that can either suppress (pro survival) or promote (pro apoptotic) apoptosis[111,112]. The pro survival subfamily includes Bcl-2, B-cell lymphoma extra-large (Bcl-XL), Bcl-w (anti-apoptotic protein), anti-apoptotic member of the Bcl-2 (A1) and myeloid cell leukemia-1 (MCL-1) whereas the pro apoptotic subfamily is classified into the multi domain group (BCL2-associated X Protein (Bax), BCL-2-antagonist/ killer (Bak) and BCL-2 related ovarian killer (Bok)) and the BCL-2 Homology 3 (BH3) only group[113].
Favorable results are being reported with the usage of inhibitors of BCL-2 and other related molecules, especially with BH3 mimetics[114,115]. Various studies suggested that apoptosis blockage is a crucial oncogenic mechanism in lymphoid malignancies and that BCL-2 overexpression is a common finding in leukemia and lymphomas, numerous antagonists of anti-apoptotic BCL-2 have been investigated and developed for the treatment of hematological neoplasms[116,117].
Santosh et al.[118] synthesized, characterized and evaluated a series of oxadiazole and hydroxypyrazoline derivatives for promising anticancer inhibitors in which compounds 13a and 13b (fig. 14) exhibited remarkable growth inhibition of MDA-MB-231 (IC50: 10.2 ±0.02 and 29.50±1.26 μM) and HT-29 (IC50: 25.91±1.12 and 20.32±1.23 μM) cell lines and flow cytometric analysis also revealed that 13a arrests both cells lines at G0/G1 phase while 13b induced G0/G1 arrest only in the HT-29 cells. Furthermore, molecular interaction studies also revealed that 13a and 13b exhibited its capacity of being a plausible Bcl-2 and CDK2 (cell division protein kinase 2) inhibitor respectively.

Figure 14: Docked structure of oxadiazole (13a) and hydroxypyrazoline (13b) derivatives
Sabour et al.[119] designed, synthesized and evaluated a new series of 3-cyanopyridine derivatives for their targeting survivin and Bcl-2/Bax. Among them, the compound 14 showed most active cytotoxic and 17.92 times more potent than 5-fluoro uracil drug. Cell cycle analysis of the most potent 14 (fig. 15) revealed that cell cycle arrest at the G2/M phase with an increase in pre G1 apoptotic cells and was subjected to apoptosis studies to estimate their apoptotic potency in which it decreases the expression of the apoptosis suppressor Bcl-2 and increases in the level of apoptosis inducer Bax. The in vitro cytotoxicity of the synthesized compound 14 was evaluated against a panel of cell lines: PC-3, HepG-2 and MDA-MB-231 with IC50 value 0.42±0.05, 1.22±0.06 and 0.66±0.03 μM respectively and in normal cell: WI-38 (Normal human fetal lung fibroblast cell line) with IC50 value 193.15±0.09 μM.

Figure 15: Docked structure of 3-cyanopyridine derivative
Topoisomerase (Topo) inhibitors:
Human DNA topoisomerase (Topo) is a nuclear enzyme and key component of the cell which can explain all topological issues associated with several vital cellular processes like DNA replication, transcription, repair, recombination, chromatin assembly and chromosome segregation[120,121]. It comprises of two subtypes Topo I and Topo II, since the discovery of Topo by J. C. Wang in 1971, have been vitally prominent molecular targets for the development of antitumor drugs[122,123]. Human Topo I make DNA single strand cleavage at a time while Topo II which is in the presence of Magnesium (Mg) (II) and adenosine triphosphate (ATP) hydrolysis, cleaves both strands of the DNA double helix to complete their catalytic functions[124,125]. Furthermore, Topo II has two isoforms: Topo IIα and Topo IIβ, both plays an essential role in chromosome segregation and proper organization[126] and these isoforms have been considered as more imperative molecular target than Topo I for designing of antitumor agents[127].
Lee et al.[128] designed, synthesized and biological evaluated a series of thiochromeno[2,3-c]quinolin-12- one derivatives for their Topo inhibition. Among them, compound 15 showed full inhibitory activities against Topo I and Topo IIα with IC50 values of 1.14±0.04, 0.22±0.04, 0.80±0.21, 0.53±0.31 and 0.53±0.08 μM in MDA-MB-231, MDA-MB-468 (Breast carcinoma cell line), MCF-7, HCT-116 and H1299 (Lung carcinoma cell line) cells respectively. Additionally recognized compound 15 as a most potent dual Topo inhibitor with low toxicity to normal cells and SAR study fig. 16) also revealed that the terminal amino group of N-2-aminoethylamino or N-3-aminopropylamino at the 6th position and 8,10-di-halogen substituents on thiochromeno[2,3-c]quinolin-12-one are crucial for the Topo inhibitions and tumor-killing activities.

Figure 16: Structure of thiochromeno [2,3-c] quinolin-12-one derivative with SAR
Jin et al.[129] designed, synthesized and evaluated a series of new quinoline and quinolinium iodide derivatives to discover potential antitumor and antibacterial agents. The compound 16 was found to be the most potent derivative with IC50 values of 4.45±0.88, 4.74±0.42, 14.54±1.96 and 32.12±3.66 μM against A549, Hela, SGC-7901 (Gastric cancer cell line) and L-02 (Liver cancer cell line) cells respectively, stronger than the positive control by 5-fluoro uracil and Methotrexate. Furthermore, compound 16 also had the most potent bacterial inhibitory activity. Additionally, the docking result suggested there were numerous crucial interactions between the 4-position of compound 16 and human Topo I, among which two main H-bonds were formed between the amino (NH) groups or aryl- O-aryl interacts with the catalytic amino acid residue of Dystroglycan (DG) 112 and Lysine (Lys) 425, respectively (fig. 17).

Figure 17: Docked structure and SAR of quinoline derivative
Tubulin polymerization inhibitors:
Microtubules are involved in numerous vital cellular functions in various eukaryotic cells such as cell growth, motility, division, intracellular trafficking and the ability to adapt several shapes to interact with the surrounding environment[130,131]. Microtubule targeting agents are a major class of tumor chemotherapeutic drugs[132,133]. Those agents bind to heterodimers of α and β tubulin which are the building blocks of cellular microtubules. There is a growing evidence displaying that interfering with microtubules normal dynamic equilibrium can engage the spindle roadblock and the arrest of cell cycle progression leading to cell death[134-136].
Jian et al.[137] were synthesized and biologically evaluated a new pyrazolo[3,4-b]pyridine bridged analogues of combretastatin A-4 possessing 3,4,5-trimethoxylphenyl groups for their anti-proliferative and tubulin polymerization inhibitory activities. Among all these analogs, the most active analogue 17 was found to induce MCF-7, MDA-MB-231, HeLa and Kyse150 (Esophageal squamous cancer cell line) cells with in vitro cytotoxicity (IC50±standard deviation (SD)) as 27.22±2.31, 27.04±6.42, 18.08±1.48 and 62.82±2.52 μM, respectively. Moreover, molecular modeling studies showed that derivative 17 most possibly occupies the colchicine site of tubulin (fig. 18).

Figure 18: Docked structure of pyrazolo [3,4-b] pyridine-bridged analog
Mirzaei et al.[138] synthesized and evaluated a series of quinoline-chalcone hybrids to discover a promising tubulin inhibitors and anticancer activity. Among all quinolones, compound 18 revealed as the most potent anti-proliferative activity with IC50 values 2.32±0.84, 2.615±0.63, 4.96±1.46, 2.32±0.46 and 4.44±1.28 against A2780 (Ovarian carcinoma cell line), A2780/RCIS (Ovarian carcinoma cell line), MCF-7, MCF-7/MX (Breast carcinoma) and HUVEC cells respectively. It is also recognized as tubulin inhibitors and induced more arrest at G2/M phase in four cancer cell lines compared to other synthesized compounds. Furthermore, SAR study, molecular dynamics simulation and molecular docking studies of compound 18 into the colchicine binding site of tubulin showed the possible interaction with the active site of tubulin (fig. 19).

Figure 19: Docked structure and SAR of quinoline-chalcone derivative
Cytochromes P450 (CYPs) inhibition:
Cytochromes P450 (CYPs) is a superfamily of hemo proteins involved in many oxidation and reduction reactions on both endogenic and xenobiotic compounds[139]. In general the CYP isoenzyme superfamily consists of 57 CYP genes and 58 pseudogenes arranged into 18 families and 43 subfamilies[140]. For instance, the CYP1 family is included of 3 members: CYP1A1 (cytochrome P450 Family 1 Subfamily A Member 1), CYP1A2 (cytochrome P450 Family 1 Subfamily A Member 2) and CYP1B1 (cytochrome P450 Family 1 Subfamily B Member 1). The latter is primarily expressed in extra hepatic mesodermal cells including steroidogenic tissues such as ovaries, testes and adrenal glands and in steroid responsive tissues such as breast, uterus and prostate[141]. In human eighteen (18) CYP gene families, including over 50 enzymes are found[139]. Despite the importance of other CYP isoforms like CYP3A4 and CYP2D6 are responsible in the metabolism of 50 % of clinical drugs[142].
Wang et al.[143] reported a class of 3-substituted 1H-pyrrolo[2,3-b]pyridine derivatives and evaluated for their in vitro anticancer activities. Among the derivatives, the optimized compound 19 displayed potent enzyme inhibition and exceptional antiproliferative effect with IC50 values from 0.109 μM to 0.245 μM on A549, MDA-MB-231 and MCF-7 cell lines. Further analysis indicated that compound 19 potently suppressed the migration of A549 cells and showed moderate inhibitory activity against various subtypes of human cytochrome P450 with percentage inhibition at 10 μM of various isozyme CYP1A2, CYP2C9, CYP2D6, CYP2C19 and CYP3A4 as 26.7, 4.9, 5.8, 37.8 and 1.3 μM, respectively (fig. 20).

Figure 20: Docked structure of 3-substituted 1H-pyrrolo[2,3-b]pyridine derivative
Mohamed et al.[144] synthesized and evaluated a new class of hetero steroids derivatives for their anticancer activities. It was observed that compounds 20a (IC50: 38.99, 28.88 and 21.98 μM) and 20b (IC50: 41.19, 31.99 and 23.91 μM) showed the highest cytotoxic effects against all tested HepG-2, Huh-7 (Liver cancer cell line) and A549 cell lines. Furthermore, molecular simulation also revealed the activity of the tested compounds against four different proteins (CDK2, CYP19 (aromatase), janus Kinase 2 and Bcl2) which are vastly implicated in tumor regulation and progression. Compound 20a and 20b found that were indicated by lowest binding energy (compound 20a=−11.3, −9.2, −11.3 and −9.8 kcal/mol and compound 20b=−10.5, −11.8, −12.0 and −9.7 kcal/mol) compared to reference ligand (fig. 21).

Figure 21: Docked structure and SAR of hetero-steroids derivatives
Miscellaneous:
Vadukoot et al.[145] reported the synthesis, SAR and biological evaluation of a series of 1H-pyrrolo[2,3-b] pyridine-2-carboxamide derivatives as selective and potent phosphodiesterase type 4B (PDE4B) inhibitors. Among all, compound 21 is a PDE4B preferring inhibitor with IC50 value 0.14 and 0.88 μM for PDE4B and PDE4D respectively and displayed acceptable in vitro absorption, distribution, metabolism and excretion (ADME) and significantly also inhibited tumour necrosis factor alpha (TNF-α) release from macrophages showed to pro inflammatory stimuli. In a radio ligand binding assay of PDE isoforms for percentage inhibition at 10 μM, compound 21 was selective against all tested isoforms with <50 % inhibition, except PDE3B with about 87 % inhibiton (fig. 22).

Figure 22: Docked structure and SAR of 1H-pyrrolo[2,3-b]pyridine-2-carboxamide derivative
Lin et al.[146] designed, synthesized and evaluated a new C-X-C chemokine receptor type 4 (CXCR4) antagonists based on an aminoquinoline template. A different potent CXCR4 antagonists were recognized, exemplified by compound 22, which revealed excellent binding affinity with CXCR4 receptor (IC50=57 nM) and inhibited C-X-C motif chemokine 12 (CXCL12) induced cytosolic calcium released (IC50=0.24 nM). Moreover, compound 22 potently inhibited CXLC12/ CXCR4 mediated cell migration in a transwell invasion assay. SAR study also revealed it as potent inhibitor, as mentioned in fig. 23.

Figure 23: Structure of aminoquinoline derivative with SAR
Xi et al.[147] designed, synthesized and evaluated a series of new 3-(thiophen-2-ylthio)pyridine derivatives to discover promising multi target antitumor agents. Distinctively compound 23 (fig. 24) with IC50 values for HepG-2 is 2.98±1.11 μM and WSU-DLCL2 (B-cell lymphoma cell line): 4.34±0.84 μM, demonstrated good inhibitory activities against Fibroblast growth factor receptor 2 (FGFR2), FGFR3, EGFR, Janus kinase and receptor originated from Nantes (RON). Additionally, the cell cycle analysis showed that compound 23 could arrest HepG-2 cells in the G1/G0 phase.

Figure 24:Docked structure of 3-(thiophen-2-ylthio)pyridine derivative
Gao et al.[148] designed, synthesized and evaluated a series of 6-substituted pyrrolo[2,3-d]pyrimidine derivatives as non-classical antifolate (dihydrofolate reductase (DHFR)) and potential anticancer inhibitors. In preliminary anti-proliferation assay of all compounds revealed submicromolar to nanomolar inhibitory effects against KB (keratin forming tumor cell line) tumor cells, whereas compounds 24a-24c also exhibited nanomolar anti proliferative activities towards SW620 (Colon cancer cell line) and A549 cells. In particular, compounds 24a-24c were considerably more potent with IC50 value for compound 24a: 0.66, 0.43 and 0.22, compound 24b: 0.78, 0.76 and 0.81 and compound 24c: 0.47, 1.52 and 0.56, against KB, SW620 and A549 cells line, respectively. The growth inhibition induced cell cycle arrest at G1 phase with S phase suppression. Moreover, molecular modeling studies also suggested two binding modes of the target compounds with DHFR (fig. 25).

Figure 25: Docked structure and SAR of 6-substituted pyrrolo[2,3-d]pyrimidine derivatives
Mizojiri et al.[149] designed, synthesized and biologically evaluated a series 1H-Pyrrolo[3,2-b] pyridine-3-carboxamide derivatives for their Acetylcoenzyme- A carboxylase1 (ACC1) inhibitory activity. Initially identified 1-methyl-1H-pyrrolo[3,2-b] pyridine-3-carboxamide derivative as a potent ACC1 inhibitor, because of it having pharmacokinetic and physicochemical issues, this led to discovery of 1-isopropyl-1H-pyrrolo[3,2-b]pyridine-3-carboxamide derivative compound 25 as a promising ACC1 inhibitor, which showed potent ACC1 inhibition as well as sufficient cellular potency with IC50 value of 150 nM against HCT-116 cells. SAR study also revealed it as potent and promising ACC1 inhibitor, as mentioned in fig. 26.

Figure 26: Structure of 1H-pyrrolo[3,2-b]pyridine-3-carboxamide derivative with SAR
Conclusion
This review study brought out pyridine as an advantageous and privileged moiety in the field of pharmaceutical sciences with its broad range of anticancer profile including numerous targets to throw enormous scientific knowledge for development and designing of newer pyridine derivatives or analogs. The thoroughgoing declaration of SAR/molecular docking will surely help the scientific society to produce effective and potent drugs with excellent pharmacological activity.
Acknowledgements:
The authors would like to thank Prof. (Dr.) Pramod Kumar Sharma, Dean, School of Medical & Allied Sciences, Galgotias University, Greater Noida (201310) for providing technical supports.
Conflict of Interests:
The authors declared no conflict of interest.
References
- Begley CG, Ellis LM. Raise standards for preclinical cancer research. Nature 2012;483:531-3.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646-74.
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61(2):69-90.
- Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Pineros M, et al. Estimating the global cancer incidence and mortality in 2018: Globocan sources and methods. International journal of cancer 2019 Apr 15;144(8):1941-53.
- WHO report on cancer: setting priorities, investing wisely and providing care for all? Geneva: World Health Organization; 2020.
- Liu Z, Delavan B, Roberts R, Tong W. Lessons learned from two decades of anticancer drugs. Trends Pharmacol Sci 2017;38(10):852-72.
- Wu XZ. A new classification system of anticancer drugs-based on cell biological mechanisms. Med Hypotheses 2006;66(5):883-7.
- Sharifnia T, Hong AL, Painter CA, Boehm JS. Emerging opportunities for target discovery in rare cancers. Cell Chem Biol 2017;24(9):1075-91.
- Pandeya SN. A textbook of medicinal chemistry. 2nd ed. Varansi: SG publisher; 2003. p. 108-13.
- Henry GD. De novo synthesis of substituted pyridines. Tetrahedron 2004;29:6043-61.
- Altaf AA, Shahzad A, Gul Z, Rasool N, Badshah A, Lal B, et al. A review on the medicinal importance of pyridine derivatives. J Drug Des Med Chem 2015;1(1):1.
- Scriven EF, Murugan R. Pyridine and pyridine derivatives Kirk-Othmer Encyclopedia of Chemical Technology. 4th ed. John Wiley & Sons. Inc: Hoboken, NJ; 2005 . p. 20.
- Ranu BC, Jana R, Sowmiah S. An improved procedure for the three-component synthesis of highly substituted pyridines using ionic liquid. J Org Chem 2007;72(8):3152-4.
- Ma X, Gang DR. The lycopodium alkaloids. Nat Prod Rep 2004;21(6):752-72.
- Jemmezi F, Kether FB, Amri I, Bassem J, Khiari JE. Synthesis and biological activity of novel benzothiazole pyridine derivatives. IOSR J Appl Chem 2014;7(1):62-4.
- Cohen ML. Changing patterns of infectious disease. Nature 2000;406:762-7.
- Sahu R, Mishra R, Majee C. An insight into primary biliary cholangitis and its recent advances in treatment: semi-synthetic analogs to combat ursodeoxycholic-acid resistance. Expert Rev Gastroenterol Hepatol 2020;14(10):985-98.
- Ibrahim HS, Eldehna WM, Abdel-Aziz HA, Elaasser MM, Abdel-Aziz MM. Improvement of antibacterial activity of some sulfa drugs through linkage to certain phthalazin-1 (2H)-one scaffolds. Eur J Med Chem 2014;85:480-6.
- Zhao LX, Sherchan J, Park JK, Jahng Y, Jeong BS, Jeong TC, et al. Synthesis, cytotoxicity and structure-activity relationship study of terpyridines. Arch Pharm Res 2006;29(12):1091-5.
- Jeong BS, Choi HY, Kwak YS, Lee ES. Synthesis of 2, 4, 6-tripyridyl pyridines and evaluation of their antitumor cytotoxicity, topoisomerase I and II inhibitory activity and structure-activity relationship. Bull Korean Chem Soc 2011;32(10):3566-70.
- El-Sharkawy KA, Ibrahim RA. New approaches for the synthesis and antitumor evaluation of pyridine, thieno [3, 4-c] pyridine, pyrazolo [3, 4-b] pyridine and pyrido [3, 4-d] pyridazine derivatives. Eur. Chem. Bull 2013;2(8):530-7.
- Shimizu S, Watanabe N, Kataoka T, Shoji T, Abe N, Morishita S, et al. Pyridine and pyridine derivatives. Ullmann's Encyclopedia of Industrial Chemistry 6th ed. New York: Wiley; 2000.
- New Drugs at FDA: CDER?s New Molecular Entities and New Therapeutic Biological Products. U.S. Food and Drug Administration; 2020.
- Cournia Z, Efstratiadis A, Kapella A, Couladouros E, Christoforidis S. WO2020039097A1; 2020.
- Cai X, Qian C, Zhai H. US20200062754A1; 2020.
- Schultz-Fademrecht C, Klebl B, Nussbaumer P, Degenhart C, Baumann M. WO2020039060A1; 2020.
- Machacek M, Witter D, Gibeau C, Huang C, Kawamura S, Sloman DL, et al. WO2020033288A1; 2020.
- Blencowe P, Charles M, Cridland A, Ekwuru T, Heald R, Macdonald E, et al. WO2020030925A1; 2020.
- Yun-Long L, Zhu W, Mei S, Glenn J. US20200040002A1; 2020.
- Strum JC, Bisi JE, Roberts PJ, Sorrentino JA. US20200022983A1; 2020.
- Sanqi Z, Ye F, Yongxiao C, Ying S, Xueyan Y, Minxing X, et al. CN110698491A; 2020.
- Castro AC, Jonaitis DT. WO2020014465A1; 2020.
- Haginoya N, Suzuki T, Hayakawa M, Ota M, Tsukadak T, Kobayashi A, et al. US20200010458A1; 2020.
- Gunzner-Toste JL, Sutherlin D, Stanley MS, Bao L, Castanedo GM, LaLonde RL, et al. US20200010420A1; 2020.
- Luzzio MJ, Freeman-Cook KD, Bhattacharya SK, Hayward MM, Hulford CA, Autry CL, et al. US20200002310A1; 2020.
- Hangauer DG, Jr. US20190367456A1; 2019.
- Ibrahim PN, Spevak W, Zhang J, Shi S, Powell B, Ma Y. US20190367507A1; 2019.
- Kumar SKC, Hood J. US20190352279A1; 2019.
- Sokolsky A, Vechorkin O, Liu K, Pan J, Yao W, Ye Q. US20190343814A1; 2019.
- Mjalli AMM, Gaddam B, Polisetti DR, Kostura MJ, Guzel M. US20190336506A1; 2019.
- Inukai T, Takeuchi J, Yasuhiro T. US20190135785A1; 2019.
- Phillipson D, Reichenbacher K, Duguid RJ, Ware JA. EP2393808B1; 2019.
- Jacobsen EJ, Blinn JR, Springer JR, Hockerman SL. US20190106439A1; 2019.
- Hood J, Wallace DM, Kumar S. RU2682245C1; 2019.
- Baloglu E, Shacham S, Senapedis W. US20180235948A1; 2018.
- Larson AM, Love K, Weight AK, Crane A, Langer RS, Klibanov AM. US20180228831A1; 2018.
- Kumar SKC, Wallace DM, Cao J, Chiruta C, Hood J. US20180228782A1; 2018.
- Hitchcock M, Mengel A, Putter V, Siemeister G, Wengner AM, Briem H, et al. US20170260198A1; 2017.
- Sherer BA, Brugger N. US20170174653A1; 2017.
- Bader B, Bone W, Briem H, Eberspacher U, Eis K, Grudzinska-Goebel J, et al. AU2015299173A1; 2017.
- Hunter KW, Crawford NP, Alsarraj J. Mechanisms of metastasis. Breast Cancer Res 2008;10(1):S2.
- Arnst JL, Hein AL, Taylor MA, Palermo NY, Contreras JI, Sonawane YA, et al. Discovery and characterization of small molecule Rac1 inhibitors. Oncotarget 2017;8(21):34586.
- Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans 2005;33(5):891-5.
- Kleer CG, Griffith KA, Sabel MS, Gallagher G, van Golen KL, Wu ZF, et al. RhoC-GTPase is a novel tissue biomarker associated with biologically aggressive carcinomas of the breast. Breast Cancer Res Treat 2005;93(2):101-10.
- Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett 2008;582(14):2093-101.
- Vlaar CP, Castillo-Pichardo L, Medina JI, Marrero-Serra CM, Velez E, Ramos Z, et al. Design, synthesis and biological evaluation of new carbazole derivatives as anti-cancer and anti-migratory agents. Bioorg Med Chem 2018;26(4):884-90.
- Litzenburger UM, Opitz CA, Sahm F, Rauschenbach KJ, Trump S, Winter M, et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014;5(4):1038.
- Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, et al. Indoleamine 2, 3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol 2011;12(9):870-8.
- Orabona C, Belladonna ML, Vacca C, Bianchi R, Fallarino F, Volpi C, et al. Cutting edge: silencing suppressor of cytokine signaling 3 expression in dendritic cells turns CD28-Ig from immune adjuvant to suppressant. J immunol 2005;174(11):6582-6.
- Ochs K, Ott M, Rauschenbach KJ, Deumelandt K, Sahm F, Opitz CA, et al. Tryptophan-2, 3-dioxygenase is regulated by prostaglandin E2 in malignant glioma via a positive signaling loop involving prostaglandin E receptor-4. J Neurochem 2016;136(6):1142-54.
- Kong KM, Zhang JW, Liu BZ, Meng GR, Zhang Q. Discovery of 5-(pyridin-3-yl)-1H-indole-4, 7-diones as indoleamine 2, 3-dioxygenase 1 (IDO1) inhibitors. Bioorg Med Chem Lett 2020;30(4):126901.
- Yang R, Chen Y, Pan L, Yang Y, Zheng Q, Hu Y, et al. Design, synthesis and structure-activity relationship study of novel naphthoindolizine and indolizinoquinoline-5, 12-dione derivatives as IDO1 inhibitors. Bioorg Med Chem 2018;26(17):4886-97.
- Tafreshi NK, Lloyd MC, Bui MM, Gillies RJ, Morse DL. Carbonic anhydrase IX as an imaging and therapeutic target for tumors and metastases. . Subcell Biochem 2014:221-54.
- Sedlakova O, Svastova E, Takacova M, Kopacek J, Pastorek J, Pastorekova S. Carbonic anhydrase IX, a hypoxia-induced catalytic component of the pH regulating machinery in tumors. Front Physiol 2014;4:400.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7(2):168-81.
- Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7(3):48.
- Kato Y, Ozawa S, Miyamoto C, Maehata Y, Suzuki A, Maeda T, et al. Acidic extracellular microenvironment and cancer. Cancer Cell Int 2013;13(1):1-8.
- Patard JJ, Fergelot P, Karakiewicz PI, Klatte T, Trinh QD, Rioux-Leclercq N, et al. Low CAIX expression and absence of VHL gene mutation are associated with tumor aggressiveness and poor survival of clear cell renal cell carcinoma. Int J Cancer 2008;123(2):395-400.
- Uslu AG, Maz TG, Nocentini A, Banoglu E, Supuran CT, Caliskan B. Benzimidazole derivatives as potent and isoform selective tumor-associated carbonic anhydrase IX/XII inhibitors. Bioorg Chem 2020;95:103544.
- Ansari MF, Idrees D, Hassan MI, Ahmad K, Avecilla F, Azam A. Design, synthesis and biological evaluation of novel pyridine-thiazolidinone derivatives as anticancer agents: targeting human carbonic anhydrase IX. Eur J Med Chem 2018;144:544-56.
- DiNardo CD, Jabbour E, Ravandi F, Takahashi K, Daver N, Routbort M, et al. IDH1 and IDH2 mutations in myelodysplastic syndromes and role in disease progression. Leukemia 2016;30(4):980-4.
- Zhou KG, Jiang LJ, Shang Z, Wang J, Huang L, Zhou JF. Potential application of IDH1 and IDH2 mutations as prognostic indicators in non-promyelocytic acute myeloid leukemia: a meta-analysis. Leuk Lymphoma 2012;53(12):2423-9.
- Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst 2010;102(13):932-41.
- Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol 2016;27(4):599-608.
- Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med 2017;9(375).
- Reitman ZJ, Parsons DW, Yan H. IDH1 and IDH2: not your typical oncogenes. Cancer cell 2010;17(3):215-6.
- Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Gorlich K, et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 2013;122(16):2877-87.
- Liu X, Hu Y, Gao A, Xu M, Gao L, Xu L, et al. Synthesis and biological evaluation of 3-aryl-4-indolyl-maleimides as potent mutant isocitrate dehydrogenase-1 inhibitors. Bioorg Med Chem 2019;27(4):589-603.
- Hu Y, Gao A, Liao H, Zhang M, Xu G, Gao L, et al. 3-(7-Azaindolyl)-4-indolylmaleimides as a novel class of mutant isocitrate dehydrogenase-1 inhibitors: Design, synthesis and biological evaluation. Arch Pharm 2018;351(10):1800039.
- Ribeiro RCJ, Kushner PJ, Baxter JD. The nuclear hormone receptor gene superfamily. Annu Rev Med 1995;46(1):443-53.
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell 1995;83(6):835.
- Kemppainen JA, Langley E, Wong CI, Bobseine K, Kelce WR, Wilson EM. Distinguishing androgen receptor agonists and antagonists: distinct mechanisms of activation by medroxyprogesterone acetate and dihydrotestosterone. Mol Endocrinol 1999;13(3):440-54.
- Dehm SM, Tindall DJ. Androgen receptor structural and functional elements: role and regulation in prostate cancer. Mol Endocrinol 2007;21(12):2855-63.
- Elshan ND, Rettig MB, Jung ME. Molecules targeting the androgen receptor (AR) signaling axis beyond the AR-Ligand binding domain. Med Res Rev 2019;39(3):910-60.
- Moras D, Gronemeyer H. The nuclear receptor ligand-binding domain: structure and function. Curr Opin Cell Biol 1998;10(3):384-91.
- Chang C, Lee SO, Yeh S, Chang TM. Androgen receptor (AR) differential roles in hormone-related tumors including prostate, bladder, kidney, lung, breast and liver. Oncogene 2014;33(25):3225-34.
- Niu Y, Chang TM, Yeh S, Ma WL, Wang YZ, Chang C. Differential androgen receptor signals in different cells explain why androgen-deprivation therapy of prostate cancer fails. Oncogene 2010;29(25):3593-604.
- Marques RB, Dits NF, Erkens-Schulze S, Van Weerden WM, Jenster G. Bypass mechanisms of the androgen receptor pathway in therapy-resistant prostate cancer cell models. PLoS One 2010;5(10):e13500.
- Perner S, Cronauer MV, Schrader AJ, Klocker H, Culig Z, Baniahmad A. Adaptive responses of androgen receptor signaling in castration-resistant prostate cancer. Oncotarget 2015;6:35542.
- Bobach C, Tennstedt S, Palberg K, Denkert A, Brandt W, de Meijere A, et al. Screening of synthetic and natural product databases: Identification of novel androgens and antiandrogens. Eur J Med Chem 2015;90:267-79.
- Ferroni C, Pepe A, Kim YS, Lee S, Guerrini A, Parenti MD, Tesei A, Zamagni A, Cortesi M, Zaffaroni N, De Cesare M. 1, 4-Substituted triazoles as nonsteroidal anti-androgens for prostate cancer treatment. J Med Chem 2017;60(7):3082-93.
- Shi YK, Wang B, Shi XL, Zhao YD, Yu B, Liu HM. Synthesis and biological evaluation of new steroidal pyridines as potential anti-prostate cancer agents. Eur J Med Chem 2018;145:11-22.
- Li D, Zhou W, Pang J, Tang Q, Zhong B, Shen C, et al. A magic drug target: androgen receptor. Med Res Rev 2019;39(5):1485-514.
- Penning TM, Wangtrakuldee P, Auchus RJ. Structural and functional biology of aldo-keto reductase steroid-transforming enzymes. Endocr Rev 2019;40(2):447-75.
- Zeng CM, Chang LL, Ying MD, Cao J, He QJ, Zhu H, et al. Aldo?keto reductase AKR1C1?AKR1C4: functions, regulation and intervention for anti-cancer therapy. Front Pharmacol 2017;8:119.
- Pippione AC, Carnovale IM, Bonanni D, Sini M, Goyal P, Marini E, et al. Potent and selective aldo-keto reductase 1C3 (AKR1C3) inhibitors based on the benzoisoxazole moiety: application of a bioisosteric scaffold hopping approach to flufenamic acid. Eur J Med Chem 2018;150:930-45.
- Zheng X, Jiang Z, Li X, Zhang C, Li Z, Wu Y, et al. Screening, synthesis, crystal structure and molecular basis of 6-amino-4-phenyl-1, 4-dihydropyrano [2, 3-c] pyrazole-5-carbonitriles as novel AKR1C3 inhibitors. Bioorg Med Chem 2018;26(22):5934-43.
- Savic MP, Ajdukovic JJ, Plavsa JJ, Bekic SS, Celic AS, Klisuric OR, et al. Evaluation of A-ring fused pyridine D-modified androstane derivatives for antiproliferative and aldo?keto reductase 1C3 inhibitory activity. MedChemComm 2018;9(6):969-81.
- Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest 2015;125(5):1780-9.
- Coussens L, Parker PJ, Rhee L, Yang-Feng TL, Chen E, Waterfield MD, Francke U, Ullrich A. Multiple, distinct forms of bovine and human protein kinase C suggest diversity in cellular signaling pathways. Science 1986;233:859-66.
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002;298:1912-34.
- Fabbro D, Jacob S, Moebitz H. The 10 things you should know about protein kinases and inhibitors. Br J Pharmacol 2015;172(11):2675-700.
- Kostler WJ, Zielinski CC. Targeting receptor tyrosine kinases in cancer. In Receptor Tyrosine Kinases: Structure, Functions and Role in Human Disease. Springer, New York, NY; 2015. p. 225-278.
- Maurer G, Tarkowski B, Baccarini M. Raf kinases in cancer?roles and therapeutic opportunities. Oncogene 2011;30(32):3477-88.
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nat Rev Cancer 2004;4(3):177-83.
- Bardelli A, Parsons DW, Silliman N, Ptak J, Szabo S, Saha S, et al. Mutational analysis of the tyrosine kinome in colorectal cancers. Science 2003;300(5621):949.
- Nawaz F, Alam O, Perwez A, Rizvi MA, Naim MJ, Siddiqui N, et al. 3'-(4-(Benzyloxy) phenyl)-1'-phenyl-5-(heteroaryl/aryl)-3, 4-dihydro-1' H, 2H-[3, 4'-bipyrazole]-2-carboxamides as EGFR kinase inhibitors: Synthesis, anticancer evaluation and molecular docking studies. Arch Pharm 2020;353(4):1900262.
- Yu YN, Han Y, Zhang F, Gao Z, Zhu T, Dong S, et al. Design, synthesis and biological evaluation of imidazo [1, 2-a] pyridine derivatives as novel PI3K/mTOR dual inhibitors. J Med Chem 2020;63(6):3028-46.
- Ullah S, El-Gamal MI, Zaib S, Anbar HS, Zaraei SO, Sbenati RM, et al. Synthesis, biological evaluation and docking studies of new pyrazole-based thiourea and sulfonamide derivatives as inhibitors of nucleotide pyrophosphatase/phosphodiesterase. Bioorg Chem 2020;99:103783.
- Meier P, Finch A, Evan G. Apoptosis in development. Nature 2000;407:796-801.
- Ghiotto F, Fais F, Bruno S. BH3-only proteins: The death-puppeteer's wires. Cytometry A 2010;77(1):11-21.
- Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 2014;15(1):49-63.
- Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007;26(9):1324-37.
- Roberts AW, Huang DC. Targeting BCL2 with BH3 mimetics: basic science and clinical application of venetoclax in chronic lymphocytic leukemia and related B cell malignancies. Clin Pharmacol Ther 2017;101(1):89-98.
- Billard C. BH3 mimetics: status of the field and new developments. Mol Cancer Ther 2013;12(9):1691-700.
- Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer 2016;16(2):99.
- Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov 2008;7(12):989-1000.
- Santosh R, Prabhu A, Selvam MK, Krishna PM, Nagaraja GK, Rekha PD. Design, synthesis and pharmacology of some oxadiazole and hydroxypyrazoline hybrids bearing thiazoyl scaffold: antiproliferative activity, molecular docking and DNA binding studies. Heliyon 2019;5(2):e01255.
- Sabour R, Harras MF, Mehany AB. Design, synthesis, cytotoxicity screening and molecular docking of new 3-cyanopyridines as survivin inhibitors and apoptosis inducers. Bioorg chem 2020;94:103358.
- Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 2002;3(6):430-40.
- Wang JC. DNA topoisomerases. Annu Rev Biochem 1996;65(1):635-92.
- Forterre P, Gribaldo S, Gadelle D, Serre MC. Origin and evolution of DNA topoisomerases. Biochimie 2007;89(4):427-46.
- Bailly C. Contemporary challenges in the design of topoisomerase II inhibitors for cancer chemotherapy. Chem Rev 2012;112(7):3611-40.
- Goto T, Wang JC. Yeast DNA topoisomerase II. An ATP-dependent type II topoisomerase that catalyzes the catenation, decatenation, unknotting and relaxation of double-stranded DNA rings. J Biol Chem 1982;257(10):5866-72.
- Pommier Y. DNA topoisomerase I inhibitors: chemistry, biology and interfacial inhibition. Chem Rev 2009;109(7):2894-902.
- McClendon AK, Osheroff N. DNA topoisomerase II, genotoxicity and cancer. Mutat Res 2007;623(1-2):83-97.
- Park S, Kadayat TM, Jun KY, Magar TB, Bist G, Shrestha A, et al. Novel 2-aryl-4-(4'-hydroxyphenyl)-5H-indeno [1, 2-b] pyridines as potent DNA non-intercalative topoisomerase catalytic inhibitors. Eur J Med Chem 2017;125:14-28.
- Lee JF, Chang TY, Liu ZF, Lee NZ, Yeh YH, Chen YS, et al. Design, synthesis and biological evaluation of heterotetracyclic quinolinone derivatives as anticancer agents targeting topoisomerases. Eur J Med Chem 2020;190:112074.
- Jin G, Xiao F, Li Z, Qi X, Zhao L, Sun X. Design, synthesis and dual evaluation of quinoline and quinolinium iodide salt derivatives as potential anticancer and antibacterial agents. ChemMedChem 2020;15(7):600-9.
- Florian S, Mitchison TJ. Anti-microtubule drugs. The Mitotic Spindle. New York: Humana Press; 2016. p. 403-21.
- Mitchison TJ. Microtubule dynamics and kinetochore function in mitosis. Annu Rev Cell Biol 1988;4(1):527-45.
- Nitika V, Kapil K. Microtubule targeting agents: a benchmark in cancer therapy. Curr Drug ther 2013;8(3):189-96.
- Risinger AL, Giles FJ, Mooberry SL. Microtubule dynamics as a target in oncology. Cancer Treat Rev 2009;35(3):255-61.
- Nepali K, Ojha R, Lee HY, Liou JP. Early investigational tubulin inhibitors as novel cancer therapeutics. Expert Opin Investig Drugs 2016;25(8):917-36.
- Mukhtar E, Adhami VM, Mukhtar H. Targeting microtubules by natural agents for cancer therapy. Mol Cancer Ther 2014;13(2):275-84.
- Rohena CC, Mooberry SL. Recent progress with microtubule stabilizers: new compounds, binding modes and cellular activities. Nat Prod Rep 2014;31(3):335-55.
- Jian XE, Yang F, Jiang CS, You WW, Zhao PL. Synthesis and biological evaluation of novel pyrazolo [3, 4-b] pyridines as cis-restricted combretastatin A-4 analogues. Bioorg Med Chem Lett 2020;30(8):127025.
- Mirzaei S, Hadizadeh F, Eisvand F, Mosaffa F, Ghodsi R. Synthesis, structure-activity relationship and molecular docking studies of novel quinoline-chalcone hybrids as potential anticancer agents and tubulin inhibitors. J Mol Struct 2020;1202:127310.
- Nebert DW, Wikvall K, Miller WL. Human cytochromes P450 in health and disease. Philos Trans R Soc Lond B Biol Sci 2013;368:20120431.
- Nelson DR, Zeldin DC, Hoffman SM, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenet Genomics 2004;14(1):1-8.
- Sutter TR, Tang YM, Hayes CL, Wo YY, Jabs EW, Li X, et al. Complete cDNA sequence of a human dioxin-inducible mRNA identifies a new gene subfamily of cytochrome P450 that maps to chromosome 2. J Biol Chem 1994;269(18):13092-9.
- Butterweck V, Derendorf H, Gaus W, Nahrstedt A, Schulz V, Unger M. Pharmacokinetic herb-drug interactions: are preventive screenings necessary and appropriate? Planta Med 2004;70(9):784-91.
- Wang R, Chen Y, Yang B, Yu S, Zhao X, Zhang C, et al. Design, synthesis, biological evaluation and molecular modeling of novel 1H-pyrrolo [2, 3-b] pyridine derivatives as potential anti-tumor agents. Bioorg Chem 2020;94:103474.
- Mohamed SA, El-Kady DS, Abd-Rabou AA, Tantawy MA, AbdElhalim MM, Elazabawy SR, et al. Synthesis of novel hybrid hetero-steroids: Molecular docking study augmented anti-proliferative properties against cancerous cells. Steroids 2020;154:108527.
- Vadukoot AK, Sharma S, Aretz CD, Kumar S, Gautam N, Alnouti Y, et al. Synthesis and SAR Studies of 1 H-Pyrrolo [2, 3-b] pyridine-2-carboxamides as Phosphodiesterase 4B (PDE4B) Inhibitors. ACS Med Chem Lett 2020;11(10):1848-54.
- Lin Y, Li Z, Xu C, Xia K, Wu S, Hao Y, et al. Design, synthesis and evaluation of novel CXCR4 antagonists based on an aminoquinoline template. Bioorg Chem 2020;99:103824.
- Xi JJ, He RY, Zhang JK, Cai ZB, Zhuang RX, Zhao YM, et al. Design, synthesis and biological evaluation of novel 3-(thiophen-2-ylthio) pyridine derivatives as potential multitarget anticancer agents. Arch Pharm 2019;352(8):1900024.
- Gao T, Zhang C, Shi X, Guo R, Zhang K, Gu J, et al. Targeting dihydrofolate reductase: Design, synthesis and biological evaluation of novel 6-substituted pyrrolo [2, 3-d] pyrimidines as nonclassical antifolates and as potential antitumor agents. Eur J Med Chem 2019;178:329-40.
- Mizojiri R, Nii N, Asano M, Sasaki M, Satoh Y, Yamamoto Y, et al. Design and synthesis of a novel 1h-pyrrolo [3, 2-b] pyridine-3-carboxamide derivative as an orally available ACC1 inhibitor. Bioorg Med Chem 2019;27(12):2521-30.





