- *Corresponding Author:
- S. K. Kashaw
Department of Pharmaceutical Sciences, Dr. Harisingh Gour University (A Central University), Sagar-470 003, India
E-mail: sushilkashaw@gmail.com
| Date of Submission | 09 March 2019 |
| Date of Revision | 13 July 2019 |
| Date of Acceptance | 11 October 2019 |
| Indian J Pharm Sci 2019;81(6):1078-1088 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
The 2,4-diaminoquinazoline is a well-known scaffold with a great potential to generate a library. In the following research work different computational strategies were applied on 2,4-diaminoquinazoline moiety to estimate its efficiency as an antitubercular scaffold among its other versatile biological activities reported in several published studies. Each data has defined specific substitution mode on the scaffold to be active on different protein site or for disease condition. The following experiment regards of 2D-QSAR, 3D-QSAR, active site and blind docking, structural orientation, pharmacophore mapping and further designing of the data set with possible real active moiety. 2D-QSAR has shown good reliability with r2 = 0.8190, q2_LOO = 0.7711 and external pred_r2 = 0.5321, along with 3D-QSAR has good predictability, q2 (r2cv) = 0.7601, pred_r2 = 0.5567. Further ligand based pharmacophore mapping was carried out for estimating atomic contribution to chemical feature for the compound. The generated hypothesis established that hydrogen accepter, donor and aromatic ring with electronegative atom are the important features. Finally, outcomes of all results were recapitulated to design new compounds. Some compounds also were designed depending on the computational finding and concludes the suitability of the scaffold to be antiTB active.
Keywords
Computational strategy, QSAR, docking, pharmacophore mapping, molecular designing, antiTB activity, validation
With the increase in the statistics of the tuberculosis (TB) affected population and the multiplicity of the types and symptoms of TB each year, a scary picture has emerged in the worldwide scenario. In 2016, 4.1 % of new cases and 19 % cases of multidrug resistant or rifampicin-resistant TB have been listed in the report[1,2]. After the resistance to isoniazid, resistance to other first line drugs is also emerging with time reducing treatment and maintenance options for TB patients. In this context, a new versatile molecular structure is the requirement for generating new leads and finally for a drug molecule development. Among different well known scaffolds, 2,4-diaminoquinazoline has been an attractive system and reported to be effective against Mycobacterium tuberculosis in a promising way[3]. The diaminoquinazoline pharmacophore is a versatile moiety, as smallest changes in the functional subgroup surprisingly revealed new novel potential compounds on a number of protein family. The moiety with different group substitution have drawn much attention due to their wide range of biological activities for example, antimalarial[4,5], antileshmanial[6], antipsychotic[7], antitubercular, anticancer drugs[8,9]. The concept of being the scaffold to be antiTB template was first found in 2000 over a public data service system[10].
In 2008 in an US patent, it was first described that the modification over the quinazoline ring system specifically at 2,4 position on the quinazoline ring as well as the benzene ring can be exploited to develop potent antiTB drug candidate[11,12]. With that goal more molecules were reported in 2015 by Odingo et al.[3] and declared the scaffold to be a promising template which should be evaluated more. In this context, computational analysis is a logical approach for the development and discovery of drug molecules in more defined way. So, it has great potentials to generate drug like lead molecules, which are constructive and futuristic approach to draw a well evaluated results in terms of mathematical expression.
Materials and Methods
QSAR study using V-life MDS:
A set of 33, 2,4-diaminoquinazoline derivatives evaluated in solid medium assay for antiTB activity reported by Odingo et al.[3] was subjected to the QSAR analysis. All QSAR studies were performed in V-Life MDS software Version 4.4. The inhibitory activity data were reported as MIC50 and converted to pMIC50 by taking negative log of MIC50 to reduce the skewness of data and activity range was taken in log 2.5 fold scatter. Table 1 shows the structure of the compounds along with their biological activity values.
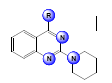 |
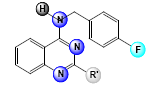 |
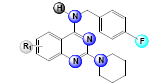 |
|||
|---|---|---|---|---|---|
| Sl. No. | R | R’ | R1 | MIC50 (µM) | log(MIC50) |
| 1 | PhCH2HN- | - | - | 9.2 | -0.96379 |
| 2 | 4-FPhCH2HN- | - | - | 7.4 | -0.86923 |
| 3 | 4-CF3PhCH2HN- | - | - | 6.6 | -0.81954 |
| 4 | 4-FPhCH2O- | - | - | 296 | -2.47129 |
| 5 | 4-FPhCH2S- | - | - | 282 | -2.45025 |
| 6 | CH3HN- | - | - | 206 | -2.31387 |
| 7 | CH(CH3)2HN- | - | - | 25 | -1.39794 |
| 8 | 4-CF3PhCH2CH2HN- | - | - | 5.7 | -0.75587 |
| 9 | 4-FPhHN- | - | - | 39 | -1.59106 |
| 10 | - | H | - | 469 | -2.67117 |
| 11 | - | Piperidin-1-yl | - | 7.4 | -0.86923 |
| 12 | - | NH2 | - | 93 | -1.96848 |
| 13 | - | CH3NH | - | 35 | -1.54407 |
| 14 | - | (CH3)2N | - | 34 | -1.53148 |
| 15 | - | Pyrrolidin-1-yl | - | 31 | -1.49136 |
| 16 | - | Piperazinyl | - | 148 | -2.17026 |
| 17 | - | (Piperidin-1-yl)CH2NH- | - | 29 | -1.4624 |
| 18 | - | H | - | 99 | -1.99564 |
| 19 | - | CH3 | - | 94 | -1.97313 |
| 20 | - | Ph | - | 76 | -1.88081 |
| 21 | - | Cyclohexyl | - | 15 | -1.17609 |
| 22 | - | - | H | 83 | -1.91908 |
| 23 | - | - | CH3 | 83 | -1.91908 |
| 24 | - | - | H | 26 | -1.41497 |
| 25 | - | - | Benzyl | 27 | -1.43136 |
| 26 | - | - | 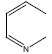 |
74 | -1.86923 |
| 27 | - | - | 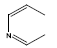 |
30 | -1.47712 |
| 28 | - | - | 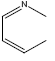 |
37 | -1.5682 |
| 29 | - | - | 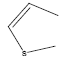 |
73 | -1.86332 |
| 30 | - | - | 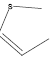 |
292 | -2.46538 |
| 31 | - | - | H | 7.4 | -0.86923 |
| 32 | - | - | 6,7-OCH3 | 25 | -1.39794 |
| 33 | - | - | - | 27.7 | -1.44248 |
Table 1: Biological Activity Data and Substituent Present In the Compounds at Different Positions
For 2D QSAR, all the compounds were subjected to energy minimization to get 3D structures, using Merck molecular force field (MMFF)[13], followed by considering distance-dependent dielectric constant of 1.0 and convergence criterion of 0.001 kcal/mol. The QSAR work sheet was generated using biological activity as dependent variable and various 2D descriptors as independent variables. The data set was divided into training and test sets were generated using 70 % search method followed by partial least squares regression coupled with stepwise forward-backward variable selection method (SW-PLS) was incorporate for estimation.
In 3D QSAR, conformers were generated by Monte Carlo conformational search method and the conformers of least energy were selected for the alignment[14]. All the compounds were aligned by template-based method, where a template was built by considering common substructures of the series. The lowest energy conformer of the most active compound was selected as a reference[15]. In the present study, all the compounds were aligned against minimum energy conformation and most active compound 10 (RMSE value 0.000110) using quinazoline nucleus as template as shown in figure 1a and the alignment of molecules is shown in figure 1b. The aligned molecules were used to generate QSAR work sheet using biological activity as dependent variable and 3D descriptors as independent variables. The data set was divided into training set and test set by considering the biological activity method along with 70 % data selection way of k-nearest neighbourmolecular field analysis coupled with stepwise forwardbackward variable selection method (known as SW kNN-MFA method) was incorporate for estimation.

Figure 1: Template structure with alignment of molecular set
(A) template structure, (B) aligned molecular set over the template and minimum energy confirmation
The pharmacophore identification studies were carried out in Mol sign module of V-life MDS 4.4 to assign the geometrical representation of the features necessary for the molecule to be active[16]. A pharmacophore model is a set of three-dimensional features that are necessary for bioactive ligands. Thus, the series of antiTB molecular set was aligned as total set and pharmacophoric was developed which contains five pharmacophoric features keeping the tolerance distance at 10 Å with maximum limit of 10.
Docking study using GOLD:
The set of 33 molecules were docked with 4 different protein molecules reported to be important for antiTB activity and the crystalline protein structures were taken from the RSCB PDB site, in order to identify the enzyme for a potent inhibitor. The chemical nature of the macromolecule and ligands greatly influence the performance of docking routines. An evaluation criterion was based on the docking scores and interacting atoms. The docking study was carried out in GOLD software version 5.2[17,18] and the ligands were prepared using Hyper Chem 7 professional. The ligand set was first subjected to MM+, then PM3 and finally through abintio quantum mechanics and the energy diagram was prepared of the whole molecular set shown in figure 2. In GOLD ChemPLP scoring method was opt with early termination off, search efficiency to be 100 % and GA run 10 000.

Figure 2: Graphical chart representation of the local energy minimization of the molecular set
Designing of a new molecule:
Depending on the results of the above mentioned studies as well as all considerations of the structural constituents of the molecular set, new molecular set was designed. The generated new molecular set first subjected to optimization and then conformers were generated by Monte Carlo conformational search method. Finally, conformers of least energy were selected for the alignment. All the compounds were aligned by template-based method with respect to the template moiety and evaluated through the 3D QSAR model prediction which estimated probable activity of the system.
Result and Discussion
In this statistical assessment, SW-PLS method was incorporated for searching the best 2D QSAR model with no randomness and biasness. The variation of the percentage selection is depicted in the Table 2. In order to construct and validate the QSAR models, both internally and externally, the data sets were divided into training (65, 70 and 75 %) and test sets (35, 30 and 25 %). Twenty nine trials were run leading to a training set with 75 % better r2, q2_LOO, pred_r2 values and F test values than the others one. The selection criteria for training and test set in a way that biological activities of all compounds in the test set lie within the maximum and minimum value of biological activities of the training set compounds. 2D QSAR Eqns were selected by optimizing the statistical results developed with the variation of descriptors in these models. The frequency of use of a particular descriptor in the population of Eqns indicated relevant contributions of the descriptors. The n-value regards the sub domain of the descriptors from the whole set incorporated. Out of the applicable domains, only 3 descriptors proved to be valuable for the model defining the chemomathematical features of the molecular set. Among the several QSAR models constructed the best regression Eqn obtained is represented in Eqn. 1, pMIC50=0.8084 (SssssCcount)–0.6067 (SulfursCount)+0.0418 (HydrogensCount)–2.4845.
| No. of trials | PLS 65% | PLS 70% | PLS 75% | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| r2 | q2 | pred_r2 | F_test | r2 | q2 | pred_r2 | F_test | r2 | q2 | pred_r2 | F_test | |
| 1 | 0.209 | 0.0713 | -1.4607 | 5.0207 | 0.6909 | 0.5579 | -0.1505 | 14.1561 | 0.5492 | 0.4051 | -0.1849 | 12.7905 |
| 2 | 0.6657 | 0.4899 | -0.9113 | 17.9202 | 0.6565 | 0.4769 | 0.2501 | 40.1372 | 0.6102 | 0.4631 | -0.1885 | 34.4327 |
| 3 | 0.6102 | 0.2886 | -2.0778 | 29.7379 | 0.2448 | 0.0805 | 0.1744 | 6.8088 | 0.4886 | 0.2313 | 0.1832 | 21.017 |
| 4 | 0.5631 | 0.4857 | -0.1351 | 24.4878 | 0.8006 | 0.7375 | 0.6994 | 25.4279 | 0.2125 | 0.0744 | 0.2145 | 5.9632 |
| 5 | 0.3447 | 0.1783 | 0.3378 | 9.9932 | 0.5754 | 0.461 | -0.9467 | 13.552 | 0.4092 | 0.2754 | -0.1116 | 15.2367 |
| 6 | 0.599 | 0.3094 | 0.4534 | 28.3782 | 0.4933 | 0.3997 | -0.3371 | 20.4475 | 0.5282 | 0.4125 | -0.8423 | 24.63 |
| 7 | 0.6451 | 0.4876 | -0.7195 | 16.3559 | 0.5876 | 0.2284 | 0.1785 | 29.9166 | 0.5466 | 0.2545 | 0.0989 | 26.5204 |
| 8 | 0.4397 | 0.0801 | -1.9336 | 14.9085 | 0.5353 | 0.4217 | -0.2859 | 24.1937 | 0.4927 | 0.2896 | 0.1657 | 21.3711 |
| 9 | 0.4831 | -0.0212 | -0.388 | 17.7568 | 0.6079 | 0.4204 | -0.108 | 32.5556 | 0.7783 | 0.6004 | -1.1625 | 23.4065 |
| 10 | 0.5419 | 0.234 | -4.0857 | 22.4788 | 0.7631 | 0.6298 | -1.9206 | 32.2131 | 0.4767 | 0.3333 | -0.2031 | 20.0431 |
| 11 | 0.7228 | 0.4987 | -2.9693 | 23.4676 | 0.6631 | 0.4701 | -1.0009 | 19.6788 | 0.743 | 0.5809 | -1.1802 | 19.2727 |
| 12 | 0.5291 | 0.2611 | 0.3135 | 21.3472 | 0.4216 | 0.0247 | -0.1828 | 15.0357 | 0.5113 | 0.1912 | 0.3482 | 23.0172 |
| 13 | 0.7031 | 0.4438 | -1.0469 | 21.3156 | 0.4869 | 0.3796 | -0.2059 | 19.9255 | 0.4605 | 0.3505 | -0.0543 | 18.781 |
| 14 | 0.4538 | 0.2931 | -0.336 | 15.7861 | 0.5029 | 0.3985 | -0.575 | 21.2489 | 0.6835 | 0.5595 | -0.1309 | 22.672 |
| 15 | 0.4788 | 0.0953 | -0.0803 | 17.4549 | 0.6426 | 0.3611 | 0.2849 | 37.7565 | 0.5302 | 0.3425 | 0.1373 | 11.8503 |
| 16 | 0.4495 | 0.3095 | 0.2345 | 15.5147 | 0.3758 | 0.0742 | 0.1395 | 12.6443 | 0.4881 | 0.3636 | -0.4036 | 20.9746 |
| 17 | 0.1549 | 0.0546 | 0.1444 | 3.4827 | 0.5569 | 0.4681 | -0.7082 | 26.3986 | 0.5966 | 0.516 | -0.1317 | 32.5334 |
| 18 | 0.6884 | 0.5218 | -0.118 | 12.5181 | 0.5869 | 0.4228 | -2.3231 | 29.835 | 0.4066 | 0.2629 | 0.2436 | 15.0124 |
| 19 | 0.604 | 0.4113 | -0.6205 | 28.9838 | 0.5602 | 0.4313 | -0.44 | 12.7364 | 0.6925 | 0.4779 | 0.1331 | 49.5506 |
| 20 | 0.6117 | 0.4613 | -0.2763 | 29.9266 | 0.7566 | 0.5128 | 0.0369 | 31.0844 | 0.5078 | 0.2945 | 0.1231 | 22.6964 |
| 21 | 0.6308 | 0.4586 | -0.2933 | 15.3771 | 0.6241 | 0.5209 | 0.1367 | 34.8703 | 0.5305 | 0.4364 | -1.056 | 24.8534 |
| 22 | 0.742 | 0.6515 | -0.4944 | 25.8789 | 0.5974 | 0.5171 | 0.052 | 31.1559 | 0.3155 | 0.2168 | -0.3956 | 10.1423 |
| 23 | 0.1823 | -0.0251 | 0.169 | 4.2364 | 0.4166 | 0.3039 | -0.453 | 14.9956 | 0.586 | 0.4791 | -0.9753 | 14.8631 |
| 24 | 0.8609 | 0.6117 | -0.5438 | 35.078 | 0.6481 | 0.21 | -0.1093 | 18.4136 | 0.5694 | 0.3055 | 0.1098 | 13.8823 |
| 25 | 0.1984 | 0.0477 | 0.222 | 4.5966 | 0.4923 | 0.39 | 0.0127 | 20.4341 | 0.6432 | 0.5519 | -0.0765 | 18.9266 |
| 26 | 0.4794 | 0.2169 | -0.9398 | 17.4992 | 0.2994 | 0.1974 | -0.1061 | 8.9763 | 0.7382 | 0.5929 | -0.0578 | 29.653 |
| 27 | 0.2878 | 0.1581 | -0.5286 | 7.6792 | 0.67 | 0.5549 | -2.5771 | 20.2989 | 0.6524 | 0.4553 | 0.2604 | 41.2847 |
| 28 | 0.6272 | 0.543 | -0.0201 | 31.9655 | 0.5556 | 0.2403 | 0.2223 | 12.5032 | 0.5331 | 0.3069 | -3.5306 | 11.9854 |
| 29 | 0.4175 | 0.2698 | -1.2741 | 13.6166 | 0.6108 | 0.4237 | -0.5184 | 15.6925 | 0.3765 | 0.2581 | 0.2926 | 13.2842 |
Table 2: Optimization for Choosing Training and Test Set By Manual Method Followed By PLS
The statistical data of the 2D QSAR model explains 81.9 % (r2 = 0.8190) of the total variance in the training set as well as it has internal (q2_LOO) and external (pred_r2) predicative ability of 77.11 and 53.21 %, respectively. The low standard error of r2_se=0.2633, q2_se=0.3321 and pred_r2se=0.2291 demonstrated accuracy of the model. The F-test=58.78 % showed the statistical significance of the model, which means that probability of failure of the model was 1 in 10 000. In addition randomization test showed confidence of 99.9 % that the generated model is not random and hence it is selected as the QSAR model.
Here, 3 descriptors, SssssCcount, SulfursCount, HydrogensCount have contributed for the estimation of 2D parameter of the QSAR model as shown in figure 3. Their effective contribution can be sustained with their correlation matrix (Table 3), which indicated that they were independent of each other, so reliability is good without the biased result. SssssCcount descriptor defines the total number of carbon connected with four single bonds and under the subclass of E-state number under the physicochemical class with positive coefficient value. It accounted for the bond formation of carbon atom within the system which can contribute to electropotential, steric hindrance as well hydrophobicity of the compounds. SulfursCount descriptor signifies number of sulphur atoms in a compound and defined subclass is element count with negative coefficient value. Sulfur atom positioned in space effects both hydrophobic and electronic orientation along with the steric effect in the molecular space by its atomic contribution to the molecular set.
| Descriptors | SssssCcount | SulfursCount | HydrogensCount |
|---|---|---|---|
| SssssCcount | 1 | ||
| SulfursCount | -0.1174 | 1 | |
| HydrogensCount | 0.1381 | 0.0730 | 1 |
Table 3: Correlation Matrix of Descriptors Used In 2d QSAR Model

Figure 3: Plots of the 2D QSAR model
(a) Contribution plot, (b) fitness plot  Training set,
Training set,  test and (c) activity plot of
test and (c) activity plot of  actual,
actual,  predicted in the 2D QSAR model
predicted in the 2D QSAR model
HydrogensCount descriptor signifies number of hydrogen atoms in a compound and it is considered under the subclass of element count with positive coefficient value. Number of hydrogen greatly affects the hydrogen bond within the system both inter and intra hydrogen bond orientation will directly affects the hydrophobicity of the molecular set.
Several 3D QSAR models were generated using SW kNN-MFA method and among these models the best model was selected on the basis of values of statistical parameters. The best SW kNN-MFA 3D QSAR models with 23 training compounds have a q2 (r2cv) value of 0.7601 and pred_r2 value of 0.5567 was considered to be the best. The kNN-MFA QSAR method explores formally the active analogue approach which implies that compounds display similar profiles of pharmacological activities. In this method the activity of each compound was predicted as average activity of k most chemically similar compounds from that data set.
In 3D QSAR studies, 3D lattice generated around 2,4-diaminouinazoline pharmacophore were used to optimize the electrostatic and steric requirements of the nucleus for antiTB activity. The generated 3D grid box, assisted in designing new compounds, which are based on the variation of the field values at the chosen points using the most active molecule and its nearest neighbour set. The points generated in SW KNN MFA 3D QSAR model are H_1014 (0.550626, 0.550762), S_669 (-0.480982, -0.473374) (figure 4a). These are the interaction energy of the set with the virtual receptor. The groups on the molecules near the lattice points need to have the interaction energy with the lattice point within the given range to be active.

Figure 4: Characte ristics of the principle pharmacophore model of the molecular set
(a) Relative positions of the local fields around aligned molecules, (b) the principle pharmacophore model of the series, (c) overlapped pharmacophore map of the molecular set, (d) the distance of the main region of the considerable points in pharmacophore
H_1014 (0.550626 0.550762), the positive range indicated that at lattice point 1014 positive hydrophobic potential is favourable for increase in the activity of the molecular set and hence a hydrophobic substituent would be favourable in that region to get more potent congeners. Alkyl group or benzene ring system as well as benzene ring substituted with appropriate functional group, which contributed to increase hydrophobic parameter will be suitable for getting more potent molecular set.
S_669(-0.480982-0.473374), the negative range indicated that at lattice point 669 negative steric potential is favourable for increase in the activity and hence less bulkier substituent is preferred in this region. Hence, bulkier group like di- or tri- substituted hetero-phenyl ring system should be removed from that particular region or else benzene ring with single substitution, hetero-phenyl ring system or hetero atomic functional group should be more preferred for better molecular lead. 2D QSAR model validation was done by estimating various statistical parameter considerations. The q2_LOO value showed the robustness of the model developed, which was within acceptable limits[19]. The Z score value of the model prepared were in acceptability range and also the residual error values were cross checked (Table 4). All the Z score values were noted when the model was prepared through the defined way and all the factors are within the acceptable limits, shown in the Table 5 which ensures the models reliability and acceptability that contributes directly to the validation of the prepared 2D QSAR model.
| 2D QSAR | 3D QSAR | |||||
|---|---|---|---|---|---|---|
| Sl. No. | Actual Activity | Predicted Activity | Residual error | Actual Activity | Predicted Activity | Residual error |
| a01 | -0.9637 | -0.9532 | -0.0105 | -0.9637 | -0.9628 | -0.0009 |
| a02 | -0.8692 | -0.8563 | -0.0129 | -0.8692 | -0.8657 | -0.0034 |
| a03 | -0.8195 | -0.81 | -0.0095 | -0.8195 | -0.8135 | -0.006 |
| a04 | -2.4712 | -2.4623 | -0.0089 | -2.4712 | -2.4709 | -0.0003 |
| a05 | -2.4502 | -2.4511 | 0.0008 | -2.4502 | -2.4496 | -0.0006 |
| a06 | -2.3138 | -2.3011 | -0.0127 | -2.3138 | -2.3299 | 0.016 |
| a07 | -1.3979 | -1.3877 | -0.0101 | -1.3979 | -1.3964 | -0.0014 |
| a08 | -0.7558 | -0.7601 | 0.0042 | -0.7558 | -0.7652 | 0.0093 |
| a09 | -1.591 | -1.5946 | 0.0036 | -1.591 | -1.587 | -0.004 |
| a10 | -2.6711 | -2.6801 | 0.0089 | -2.6711 | -2.6707 | -0.0003 |
| a11 | -0.8692 | -0.8566 | -0.0125 | -0.8692 | -0.8594 | -0.0098 |
| a12 | -1.9684 | -1.9597 | -0.0087 | -1.9684 | -1.9589 | -0.0095 |
| a13 | -1.544 | -1.534 | -0.01 | -1.544 | -1.5435 | -0.0005 |
| a14 | -1.5314 | -1.5296 | -0.0018 | -1.5314 | -1.5298 | -0.0015 |
| a15 | -1.4913 | -1.4895 | -0.0018 | -1.4913 | -1.4876 | -0.0036 |
| a16 | -2.1702 | -2.1689 | -0.0013 | -2.1702 | -2.1699 | -0.0002 |
| a17 | -1.4624 | -1.4589 | -0.0035 | -1.4624 | -1.4269 | -0.0354 |
| a18 | -1.9956 | -1.9907 | -0.0049 | -1.9956 | -1.9896 | -0.0059 |
| a19 | -1.9731 | -1.9574 | -0.0153 | -1.9731 | -1.9685 | -0.0045 |
| a20 | -1.8808 | -1.875 | -0.0052 | -1.8808 | -1.8879 | 0.0071 |
| a21 | -1.176 | -1.178 | 0.0019 | -1.176 | -1.175 | -0.001 |
| a22 | -1.919 | -1.918 | -0.001 | -1.919 | -1.9176 | -0.0014 |
| a23 | -1.919 | -1.918 | -0.001 | -1.919 | -1.9176 | -0.0013 |
| a24 | -1.4149 | -1.4175 | 0.0026 | -1.4149 | -1.4124 | -0.0025 |
| a25 | -1.4313 | -1.4299 | -0.0014 | -1.4313 | -1.4286 | -0.0027 |
| a26 | -1.8692 | -1.7458 | -0.1234 | -1.8692 | -1.859 | -0.0102 |
| a27 | -1.4771 | -1.478 | 0.0008 | -1.4771 | -1.4768 | -0.0002 |
| a28 | -1.5682 | -1.5548 | -0.0133 | -1.5682 | -1.5563 | -0.0118 |
| a29 | -1.8633 | -1.8544 | -0.0089 | -1.8633 | -1.8754 | 0.01215 |
| a30 | -2.4653 | -2.4588 | -0.0065 | -2.4653 | -2.4498 | -0.0155 |
| a31 | -0.8692 | -0.8547 | -0.0145 | -0.8692 | -0.8447 | -0.0244 |
| a32 | -1.3979 | -1.3844 | -0.0134 | -1.3979 | -1.3897 | -0.0081 |
| a33 | -1.4424 | -1.4475 | 0.005 | -1.4424 | -1.4397 | -0.0027 |
Table 4: Residual Error Values of 2d QSAR and 3d QSAR Models
| Parameters | 2D QSAR model |
|---|---|
| Z-Score R2 | 7.3005 |
| Z-Score Q2 | 6.7067 |
| Best Rand R2 | 0.5285 |
| Best Rand Q2 | 0.39 |
| Alpha Rand R2 | 0 |
| Alpha Rand Q2 | 0.001 |
| Z Score Pred R2 | 2.4511 |
| Best Rand Pred R2 | 0.4805 |
| Alpha Rand Pred R2 | 0.001 |
Table 5: Validation Parameters of the 2d QSAR Model
For 3D QSAR, the residual error is given in Table 4. The actual vs. predicted and vice versa were plotted to estimate the reliability[20-21] of the 3D QSAR model’s predictability. The high value of cross validation parameter did not ensure reliable predictability of the model which was the key feature of a QSAR model[22]. Here, R² of the two regression lines were equal and the slope values have differed by less than 0.1 which ensured the reliability of the model prepared shown in the figure 4b. The residual error of each compound was less than 0.01 which the acceptable limit and confirmed that the model has a predictability of less error chance and failure.
Pharmacophore is a 3D description of the features needed for activity, which included hydrogen bond donors, hydrogen bond acceptors, aromatic ring, hydrophobic groups, electron donating group, electron accepting groups, which finally contributed to the chemical features of the moiety to be active. A set of pharmacophore hypothesis was generated using Mol Sign, each of the set contained the following features, hydrogen bond acceptors (blue colored cage), aromatic ring (yellow colored cage) and electronegative group (orange colored cage). The ideal bond distance of the crucial points in the structure was noted in Table 6 and figure 4c. The 1 and 3 positioned nitrogen atoms of the quinazoline ring system are important feature, which can generate electron donor and hydrogen bond acceptor regions. The benzene ring systems are useful as the electron cloud rich points for nucleophilic interaction and the electronegative fluorine substitution over the 4-aminophenolate group at position 4 of the quinazoline ring system is another crucial point for interaction as electronegative atomic region. The overlapped phamacophore map of the whole molecular set also concluded the same chemical features shown in figure 4d. The pharmacophoric features explored here are very much significant in contributing potent activity; these were considered for designing the new molecular set.
| 1st Point | 2nd Point | Distance (Ǻ) |
|---|---|---|
| N1 (Hydrogen acceptor1) | N2 (Hydrogen acceptor2) | 2.389 |
| N1 (Hydrogen acceptor1) | Aromatic ring | 6.304 |
| N1 (Hydrogen acceptor1) | Electronegative region | 8.104 |
| N2 (Hydrogen acceptor2) | Aromatic ring | 4.350 |
| N2 (Hydrogen acceptor2) | Electronegative region | 6.489 |
| Aromatic ring | Electronegative region | 2.504 |
Table 6: Bond Distance Details of the Pharmacophoric Regional Points
The probable mechanism of action of the 2,4-diaminoquinazoline moiety has not been well defined or being reported, only has been reported to be antiTB activity in defined medium against virulent strain of M. tuberculosis. A set of the reported proteins for antiTB purpose was used for the docking to investigate the probable interacting points and the active site in macromolecule. The following proteins were under taken for docking study: enoyl ACPreductase (PDB ID 1ZID, 2IDZ)[23,24], caseinolytic proteases (PDB ID 2CE3)[25], malate synthase (PDB ID 3S9I)[26], MmpL membrane protein (PDB ID 4Y0L)[27]. The macromolecules are reported to have active sites for the antiTB molecules as well as can be serve as attractive drug target for the development of structure based drug design. Docking procedure includes both active site docking and blind docking. Only those water molecules were considered which are present within the active site. The procedure was carried out using Hermes 5.2 software. In case of blind docking no water molecules were considered as it was exceeding the limit of number of water molecule extracted by the software (i.e., 25 molecules).
Active site docking is performed with enoyl ACP reductase enzyme while caseinolytic proteases, MmpL membrane protein were subjected for the blind docking. But the molecular set of 2,4-diaminoquinazoline neither shown good docking score nor any hydrogen bond interaction was observed at the reported active site as well as in case of blind docking. Its interactive protein doesn’t inlcude the list used and demands for more research which open up for more possible ideas.
Validation of the method was done using rescoring method for the blind docking and for the active site docking or direct docking superimpose fitting method using Pymol 2.0.7. The scoring vs rescoring of the total molecular set and vice versa were plotted to estimate the reliability of the docking method used. The R² of the two regression lines are equal and the slope values have differed by less than 0.1, which ensured the reliability of the method. On the other side, the fitting was done over 42 atoms of the 81 atoms constituted co-crystal system extracted from the reported pdb file over the docked conformer co-crystal system, which contributes over 50 % of the atom fitted within 2Ǻ of acceptability range (figure 5a).

Figure 5: Molecular docking illustration
(a) Superimposition of the macromolecular co-crystal reported to the docked, (b) optimized structure of the most active predicted molecule
The results of 2DQSAR such as SssssCcount, SulfursCount and hydrogen count, pointed towards the hydrophobicity and steric potential of the molecules, which strongly supported the results of 3D QSAR.
So, an optimum balance of hyrdophobicity and steric potential distribution is required in the molecular system for developing better moieties. For the development of better molecular set, the following major points of the QSAR results was considered; instead of huge bulky group, substitution was done with those functional groups which contributed hydrophobicity to the whole molecular system with less steric hindrance, such as, benzene ring, heterocyclic ring, hetero-alicyclic ring with very less further substitution on them. Among the molecular set, compound 10 was reported to be the most active molecule (reported MIC50= 469, -log MIC50= -2.6707), which was considered as pharmacophoric reference moiety. Taking all considerations, a set of 1000 molecular system was generated. The activity of designed compounds was predicted using the developed 3D QSAR model. Only the good and efficient molecule having activity greater than the reported value is reported in Table 4.
The most active compound predicted has all the quality ensured in the result of the pharmacophore and the QSAR results. Its basic template is 2,4-diaminoquinazoline ring with substitution at amino groups with piperidine and 4-aminophenolate group over the other amino position. This type of compound has nowhere being reported nor being evaluated. In our study it has estimated to be a potent moiety and structurally is possible to be a real molecule.
The molecular structure was subjected to the MM+ method then, PM3 and finally using DFT b3lyp/6-311g method. So the optimized structure (figure 5b) of the designed molecule is a stable and real possible molecule. The IR spectrum given in figure 6, is the ideal one, which provides about the functional groups present in the system. This concluded the molecule is not an arbitrary outcome and imaginary, but could exist in reality. So, further development of the molecular set could reveal whether this moiety could be a drug like molecule or further molecular set development will be required for more suitable pharmacologically active molecule.

Figure 6: Ideal IR spectrum of the designed molecule
This scaffold was indicated in a number of studies for various activities and established to be a potent lead molecule. The notable reports were cited here to summaries the importance of the scaffold and also the small variations that developed moiety to be an attractive one. A set of substituted 2,4 diaminoquinazoline reported to be antimalarial in which derivatives were prepared by substitution on both the benzenoid ring system and 2 and 4 positioned amino groups with piperizine or pyridine or closed heterocyclic ring systems simultaneously. It revealed that these small variation in the substituents and different combination of substitutions over the positions on the basic structure would lead to biologically active moieties[4].
Hao et al.[8] focused on anticancer activity of the scaffold on PAK4 protein, which is a cancer pathway protein family in which N2 and N4 substituted with either 1H-indazol-5-yl or, (1H-indol-3-yl)ethyl simultaneously with the substitution at the benzenoid ring with methoxy or halogen substitution at different position turning the basic scaffold to be a PAK4 inhibitor molecule. Horn et al.[28] reported that substitution at the N2 and N4 either with benzyl or phenyl simultaneously produced molecular set with different permutations and combinations with antibacterial activity. Besides, branched chain substitution on same position has played some role along with some more moieties were reported which were substituted at the quinazoline benzenoid ring of the system; leading to make the scaffold active against multi-drug resistant Stephylococcus aureus[28]. 4-substituted-piperazine-1-carbodithioate derivatives of 2,4-diaminoquinazoline have been reported by Cao et al., who screened those against a panel of five human tumour cell lines, A549, MCF-7, HeLa, HT29 and HCT-116 for antitumor activity and the results clearly depicted a molecular set from the scaffold to be antitumor[29].
The 2,4-diaminoquinazoline is a β-catenin inhibitor, which could disrupt the transcriptional activity of β-catenin and ultimately interfere many of the downstream genes expression. TGFβ-induced PPARγ expression was significantly inhibited by 2,4-diaminoquinazoline, which clearly demonstrated that β-catenin was an upstream regulator of PPARγ and critical for its expression[30]. The 7-(benzimidazol-1-yl) substituted derivatives of 2,4-diaminoquinazoline are DHFR inhibitors, among them compounds containing thiazol-2-yl group in the 2-position of the benzimidazole is highly potent in case of S. aureus DHFR as well as highly selective[31]. Chao et al. reported the scaffold to be active against dengue virus. The new derivatives were prepared by the substitution on the benzenoid ring at position 5,6,8 mainly with methoxy or OPh group substitution, i.e., viral target has a spatially restricted conformation that accommodates a narrow range of C-5 substituent[32].
A molecular set of substituting the amino group and benzenoid ring has been reported as antileishmanial scaffold. In some moieties the di-amino same group substitution with the benzenoid substitution over C5 and C6 positions have produced active moiety and some are outcome of the multiple substitution on the benzenoid ring system[33]. A report of 2017 revealed the potent antibacterial activities of N2- benzyl-N4-methylquinazoline-2,4-diamines against Gram-negative A. baumannii and multidrug resistant infectious species. The 6 or 7 substituted molecules displayed promising activity, with MICs ranging from 0.5 to 30 μM. The most potent in vitro activities were obtained with quinazoline-2,4-diamines bearing a N2- benzyl moiety and a N4-methyl group[34].
A well-defined protocol of computational experiments were followed in the present study, which managed to reach a conclusion that 2,4-diaminoquinazoline scaffold has high potential to be developed as a lead molecular set which could be subjected to lead optimization. In accordance to QSAR analysis an optimum balance of hydrophobicity and steric potential distribution is required in the molecular system for better moieties. Similar findings were observed in pharmacophore mapping, where 1 and 3 positioned nitrogen atoms of the quinazoline ring are important along with the benzene ring and the electronegative fluorine substitution over the 4-aminophenolate group at position 4 of the quinazoline ring system. Designing and evaluation of the activity through 3D QSAR model revealed that the substitution of piperidine ring at 2-amino position and 4-aminophenolate group over the 4-amino position is prime requirement as observed from QSAR and pharmacophore mapping results. The final molecule reported included all the features estimated and believed to be an active moiety. Possibly larger library generation might be required for identifying a potential candidate drug molecule. As the biological cell interaction functionality has not been checked in a conventional way, but has been checked through the protein interaction docking method, a huge probability existed that in the long run a compound could be identified, which could serve as a potential antiTB drug.
Acknowledgement:
One of the authors PB wishes to thanks AICTE for the grant of fellowship.
Conflict of interest:
The authors confirm that this article content has no conflict of interest.
References
- Who TB sheet for drug resistant TB [cited 2018 March 24]. Available from: http://www.who.int/tb/areas-of-work/drug-resistant-tb/en.
- WHO Global Tuberculosis report 2017 [cited 2018 February 21]. Available from: http://www.who.int/tb/publications/globalreports /en.
- Odingo J, Malley T, Kesicki EA, Alling T, Bailey MA, Early J, et al. Synthesis and evaluation of the 2,4-diaminoquinazoline series as anti-tubercular agents. Bioorg Med Chem 2014;22:6965-79.
- Sundriyal S, Malmquist NA, Carona J, Blundelld S, Liue F, Chene X, et al. Development of Diaminoquinazoline Histone Lysine Methyltransferase Inhibitors as Potent Blood-stage Anti-Malarial Compounds. Chem Med 2014;9(10):2360-73.
- Mishra M, Mishra VK, Senger P, Pathak AK, Kashaw SK. Exploring QSAR studies on 4-substituted quinazoline derivatives as antimalarial compounds for the development of predictive models. Med Chem Res 2014;23:1397-405.
- Zhu X, Horn KSV, Barber M, Yang S, Wang MZ, Manetsch R, et al. SAR refinement of antileishmanial N 2,N 4-disubstituted quinazoline-2,4-diamines. Bioorg Med Chem 2015;23(16):5182-9.
- Mohamed T, Shakeri A, Tin G, Rao PPN. Structure−Activity Relationship Studies of Isomeric 2,4-Diaminoquinazolines on β-Amyloid Aggregation Kinetics. ACS Med Chem Lett 2016;7:502-7.
- Hao C, Huang W, Li X, Guo J, Chen M, Yan Z, et al. Development of 2, 4-diaminoquinazoline derivatives as potent PAK4 inhibitors by the core refinement strategy. Eur J Med Chem 2017;131:1-13.
- Lin LC, Hsu SL, Wu CL, Hsueh CM. TGFβ can stimulate the p38/b-catenin/PPARc signaling pathway to promote the EMT, invasion and migration of non-small cell lung cancer (H460 cells). Clin Exp Metastasis 2014;31:881-95.
- Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, et al. Drug Bank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res 2008;36:D901-6.
- Wynne G, DeMoor O, Johnson P, Vickers R, inventor; Infectious Disease Research Institute, assignee. Use of Compounds for Preparing Anti-tuberculosis Agents. WPO 20100317607. 2008 Dec 16.
- Ananthan S, Faaleolea ER, Goldman RC, Hobrath JV, Kwong CD, Laughon BE, et al. High-throughput screening for inhibitors of Mycobacterium tuberculosis H37Rv. Tuberculosis 2009;89(5):334-53.
- Halgren TA. Merck molecular force field. III. Molecularn geometries and vibrational frequencies for MMFF94. J Comput Chem 1996;17:553-86
- Ajmani S, Jadhav K, Kulkarni SA, Three-Dimensional QSAR Using the k-Nearest Neighbor Method and Its Interpretation. J Chem Inf Model 2006;46:24-31.
- Xu M, Zhang A, Han S, Wang L. Studies of 3d-quantitative structure–activity relationships on a set of nitroaromatic compounds: CoMFA, advanced CoMFA and CoMSIA, Chemosphere 2002;48:707-15.
- Vitthal UB, Pratapro GS. 3D QSAR and pharmacophore modeling on substituted cyanopyrrolidines as type-II anti-diabetic agents potential dipeptidyl peptidase-IV Inhibitors. Pharmacophore 2016;7(5):342-8.
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727-48.
- Verdonk ML, Cole JC, Hartshorn HJ, Murray CW, Taylor RD. Improved Protein-Ligand Docking using GOLD. Proteins 2003;52:609-23.
- Gramatica P. Principles of QSAR models validation: internal and external. QSAR Comb Sci 2007;26(5):694-701.
- Zheng W, Tropsha A. Novel Variable Selection Quantitative Structure-Property Relationship Approach Based on the k-Nearest-Neighbor Principle. J Chem Inf Comput Sci 2000;40:185-94
- Shen M, Xiao Y, Golbraikh A, Gombar VK, Tropsha A. Development and Validation of k-Nearest-Neighbor QSPR Models of Metabolic Stability of Drug Candidates. J Med Chem 2003;46:3013-3020.
- Golbraikh A, Tropsha A. Beware of q2. J Mol Graphics Model 2002;20:269-76.
- Rozwarski DA, Grant GA, Barton DH, Jacobs WR, Sacchettini JC. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 1998;279(5347):98-102.
- Dias MV, Vasconcelos IB, Prado AM, Fadel V, Basso LA, Azevedo WF, et al. Crystallographic studies on the binding of isonicotinyl-NAD adduct to wild-type and isoniazid resistant 2-trans-enoyl-ACP (CoA) reductase from Mycobacterium tuberculosis. J Struct Biol 2007;159(3):369-80.
- Ingvarsson H, Maté MJ, Högbom M, Portnoï D, Benaroudj N, Alzari PM, et al. Insights into the inter-ring plasticity of caseinolytic proteases from the X-ray structure of Mycobacterium tuberculosis ClpP1. Acta Crystallogr D Biol Crystallogr 2007;63(2):249-59.
- Krieger IV, Freundlich JS, Gawandi VB, Roberts JP, Gawandi VB, Sun Q, et al. Structure-guided discovery of phenyl-diketo acids as potent inhibitors of M. tuberculosis malate synthase. Chem Biol 2012;19(12):1556-67.
- Chim N, Torres R, Liu Y, Capri J, Batot G, Whitelegge JP, Goulding CW. The Structure and Interactions of Periplasmic Domains of Crucial MmpL Membrane Proteins from Mycobacterium tuberculosis. Chem Biol 2015;22(8):1098-107.
- Horn KSV, Burda WN, Fleeman R, Shaw LN, Manetsch R. Antibacterial Activity of a Series of N2,N4-Disubstituted Quinazoline-2,4-diamines. J Med Chem 2014;57(7):3075-93.
- Cao SL, Han Y, Yuan CZ, Wang Y, Xiahou ZK, Liao J, et al. Synthesis and antiproliferative activity of 4-substituted-piperazine- 1-carbodithioate derivatives of 2,4-diaminoquinazoline. Eur J Med Chem 2013;64:401-9.
- Chen Z, Venkatesan AM, Dehnhardt CM, Dos Santos O, Delos Santos E, Ayral-Kaloustian S, et al. 2,4-Diamino-quinazolines as inhibitors of β-catenin/Tcf-4 pathway: potential treatment for colorectal cancer. Bioorg Med Chem Lett 2009;19:4980-3.
- Lam T, Hilgers MT, Cunningham ML, Kwan BP, Nelson KJ, Driver VB, et al. Structure-Based Design of New Dihydrofolate Reductase Antibacterial Agents: 7-(Benzimidazol-1-yl)-2,4-diaminoquinazolines. J Med Chem 2014;57:651-68.
- Chao B, Tong XK, Tang W, Li DW, He PL, Garcia JM, et al. Discovery and Optimization of 2,4-Diaminoquinazoline Derivatives as a New Class of Potent Dengue Virus Inhibitors. J Med Chem 2012;55:3135-43.
- Horn KSV, Zhu X, Pandharkar T, Yang S, Vesely B, Vanaerschot M, et al. Antileishmanial Activity of a Series of N2,N4-Disubstituted Quinazoline-2,4-diamines. J Med Chem 2014;57(12):5141-56.
- Fleeman R, Horn KSV, Barber MM, Burda WN, Flanigan DL, Manetsch R, et al. Characterizing the Antimicrobial Activity of N2,N4-Disubstituted Quinazoline-2,4-Diamines Towards Multidrug Resistant Acinetobacter baumannii. Antimicrob Agents Chemother 2017;61(6):59-17.





