- *Corresponding Author:
- Neelam Seedher
Department of Chemistry, Panjab University, Chandigarh-160 014, India
E-mail: nseedher@yahoo.com
| Date of Submission | 03 March 2005 |
| Date of Revision | 22 August 2005 |
| Date of Acceptance | 19 May 2006 |
| Indian J Pharm Sci, 2006, 68 (3): 327-331 |
Abstract
Mechanism of interaction of three isoxazolyl penicillins, cloxacillin sodium, dicloxacillin sodium, and flucloxacillin sodium - with bovine serum albumin has been studied using fluorescence spectroscopic technique. The stoichiometry of the interaction was found to be 1:1, and association constants were of the order of 10 4sub in each case. The nature of drug-protein interaction could be predicted from the thermodynamic parameters for the binding. High positive entropy changes and positive enthalpy changes indicated that hydrophobic interactions are predominantly involved in the binding of these drugs to serum albumin. Binding studies carried out in the presence of hydrophobic probe 1-anilinonaphthalene-8-sulfonate (ANS) showed that the drugs and ANS do not share a common site on the albumin molecule. Stern-Volmer analysis of the fluorescence data showed that both the tryptophan residues of albumin are involved; but they are not fully accessible to the drugs, and static quenching mechanism is operative.
β-lactam antibiotics contain a highly strained and reactive cyclic amide and are susceptible to chemical and enzymatic degradation. Acids, bases, and enzyme β-lactamase directly attack the beta lactam bond and inactivate the antibiotic [1]. β-lactamases are not particularly tolerant to steric hindrance when it occurs at the side chain amide. The antibiotics cloxacillin, dicloxacillin, and flucloxacillin in which a substituted isoxazolyl ring system is used as a bioisosteric replacement for the benzyl group in penicillin G are β-lactamase-resistant semi-synthetic penicillins [2]. They are also sufficiently acid-stable to be taken orally [3].
Isoxazolyl penicillins are highly bound to serum proteins, mainly albumin [4], and therefore, small changes in binding have a large effect on the proportion of free pharmacologically active drug in plasma. Thus, the nature and magnitude of the interaction of penicillins with serum albumin has important pharmacokinetic and pharmacodynamic implications and is a determinant factor in their therapeutic properties [5-6]. High serum protein binding has several other major effects, including restricted tissue distribution, reduced penetration into interstitial spaces and inflammatory fluids, delayed elimination, and interference with biological activity [7].
Barbosa et al. [8-9] and others [10] have used a range of physicochemical properties such as solution conductivity, electrokinetic behaviour, partial specific volume and adiabatic compressibility to study the interaction of cloxacillin and dicloxacillin with human serum albumin. Some work on the binding of isoxazolyl penicillins with serum albumin using microcalorimetric and ultrafiltration technique has also been reported [11-12]. Although these drugs are being frequently prescribed, no recent detailed study on the mechanism of binding of isoxazolyl penicillins with serum albumin using spectroscopic methods is available. In the present paper, binding parameters and the nature of forces involved in the interaction of three drugs cloxacillin sodium, dicloxacillin sodium and flucloxacillin sodium with bovine serum albumin has been studied using fluorescence spectroscopic technique. Human and bovine serum albumin exhibit similar binding chemistry due to the high percentage of sequence identities between the two proteins [13].
Materials and Methods
Pure drug samples (cloxacillin sodium, dicloxacillin sodium, and flucloxacillin sodium) were obtained as gift from various manufacturers. Serum albumin, bovine (BSA), and fluorescent probe 1-anilinonaphthalene-8-sulfonate (ANS) were purchased from Sigma Chemical Co., St. Louis, U.S.A. All other reagents were of analytical grade. Water used was double distilled in all-glass apparatus. BSA solutions were prepared based on molecular weight of 66,000. All experiments were carried out in 0.10 M phosphate buffer using fluorescence spectroscopic technique. Perkin Elmer fluorescence spectrophotometer (MPF 44B) equipped with a 150 W xenon lamp source was used.
Determination of binding parameters
For the determination of binding parameters, 2 ml of 10 μM albumin solution was taken in a quartz cell and increasing amounts of drug stock solution (500 μM) was added. Albumin concentration was kept fixed at 10 μM by adding the same volume of 20 μM albumin to the cell. Fluorescence spectra were recorded in the range of 280400 nm after excitation at 295 nm, in each case. Intrinsic fluorescence of protein was measured; drugs used did not have any fluorescence at the emission wavelength of protein. No correction for inner filter effect was applied since the drugs had very low absorbance (less than 0.05) at the excitation and emission wavelengths.
Stoichiometry of the drug-protein interaction was determined by the method of continuous variations [14-15] The fluorescence change (ΔF = Fprotein+drug-Fprotein ) of a series of protein-drug mixtures was measured under such conditions that the total concentration of ‘drug plus protein’ was held constant at 10 μM, but the respective mole fraction of each was varied. ΔF was plotted against the mole fraction of drug (Job’s plot), and the stoichiometry of binding was obtained from the maximum in the plot, in each case.
Data analysis
Data was analysed as follows using Ward method [16]. The fractional occupancy of the total protein binding sites by drug was obtained from the ratio θ = ΔF/ΔFmax [17-18] where ΔF = F0-F, F0 and F are the fluorescence intensities of serum albumin in the absence and presence of drug, respectively. ΔFmax values were obtained from the double reciprocal (1/ΔF versus 1/Dt) plots.
If Pt is the total protein concentration and n is the number of binding sites, the total number of sites on protein is given by nPt, and the concentration of bound sites on protein is given by nθPt [16], which is also equal to the concentration of the bound drug (Db). Df, the number of moles of free drug, was obtained from the difference Dt-Db, where Dt is the total drug added. The amount bound was expressed as moles of drug bound per mole of protein, (r = Db/Pt). The binding parameters were computed directly by fitting the experimental data (r and Df values) to the following general equation (Scatchard equation) using an iterative non-linear least squares regression program developed for this purpose:
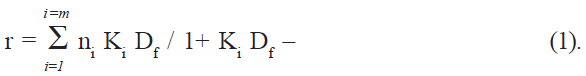
The association constant (K) and the number of binding sites (n) were determined at three different temperatures (10, 17, and 27°), and the thermodynamic parameters for binding were calculated using equations ΔG° = -RT ln K– (2) and log K = - ΔH°/2.303RT + ΔS°/2.303R – (3).
Binding studies in the presence of hydrophobic probe ANS
Experiments were also carried out in the presence of hydrophobic probe ANS [19-20]. In the first set of experiments, interaction of drugs and ANS with albumin was studied under identical conditions. Albumin concentration was kept fixed at 10 μM and ANS/drug concentration was varied from 2 to 15 μM. Fluorescence of albumin was recorded in the range 280-400 nm after . excitation at 295 nm. In the second set of experiments, albumin-ANS interaction was studied in the presence and absence of increasing concentrations of the drug. The fluorescence of ANS was recorded in the range 350 to 500 nm after excitation at 370 nm. Increasing amounts of drug was added to an equimolar albumin-ANS mixture (10 μM each). The drug concentration was varied from 10 μM to 75 μM and the concentration of albumin-ANS mixture was kept fixed at 10 μM each by adding the same volume of albumin-ANS mixture (20 μM each) to the cell. The percentage displacement of probe (D) was calculated using the relationship D = (F1 - F2)/F1 × 100, where F1 and F2 are the fluorescence intensities of ANS in the absence and presence of drug, respectively.
Results and Discussion
Bovine serum albumin (BSA) represents 52-60% of the total plasma protein and is the major binding protein for anionic drugs. BSA consists of a chain of 580 amino acid residues forming a single polypeptide that contains three domains (I-III) and two tryptophan residues, located at position 134 in subdomain IB and position 212 in subdomain IIA [21]. Intrinsic fluorescence of BSA was measured by selectively exciting the tryptophan residues at 295 nm. Cloxacillin, dicloxacillin, and flucloxacillin are structurally similar. Dicloxacillin differs from cloxacillin by the presence of an additional chlorine atom, and flucloxacillin by the presence of a fluorine atom, on the aromatic ring in the acylamino side chain. They are anionic molecules with pKa values in the range 2.7-2.8 and will be fully ionised in aqueous solution at pH 7.4 [10].
All the three drugs were found to quench the intrinsic fluorescence of serum albumin. However, there was no observable shift in the wavelength for maximum emission. The stoichiometry of the interaction was determined by the method of continuous variations, described in the experimental section. The maximum in the fluorescence change in Job’s plot occurred at 0.5 mole fraction of drug, corresponding to 1:1 stoichiometry, in each case. The Job’s plot for dicloxacillin-BSA system is shown in fig. 1. The percentage of drug bound [(D/D) x 100] at different drug:protein ratios was calculated from the data and is given in Table 1. The values were found to decrease with increase in the drug:protein ratio. At low drug:protein ratios, a significant fraction of the added drug was bound in each case. It may be mentioned that low drug:protein ratios are frequently encountered in the physiological system since in blood, the serum albumin concentration is very large (0.53-0.75 mM).

Fig. 1: Job’s plot for dicloxacillin-BSA system.
| Drug/Protein ratio (Dt/Pt) | % drug bound | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cloxacillin | Dicloxacillin | Flucloxacillin | |||||||
| 283.15 | 290.15 | 300.15 | 283.15 | 290.15 | 300.15 | 283.15 | 290.15 | 300.15 | |
| 0.5 | 36.01 | 49.43 | 53.42 | 48.51 | 48.69 | 52.98 | 37.18 | 41.83 | 52.36 |
| 1.0 | 19.42 | 26.24 | 27.97 | 25.73 | 26.84 | 29.05 | 19.72 | 22.03 | 27.46 |
| 1.5 | 13.87 | 18.49 | 19.47 | 18.11 | 19.55 | 20.37 | 13.91 | 15.44 | 19.18 |
| 2.0 | 11.13 | 14.64 | 15.25 | 14.34 | 15.90 | 17.08 | 10.96 | 12.13 | 15.02 |
| 2.5 | 9.48 | 12.33 | 12.71 | 12.06 | 13.73 | 13.94 | 9.22 | 10.14 | 12.52 |
| 3.0 | 8.35 | 10.76 | 10.99 | 10.54 | 12.26 | 13.09 | 8.08 | 8.80 | 10.86 |
| 3.5 | 7.56 | 9.66 | 9.79 | 9.46 | 11.23 | 11.17 | 7.21 | 7.86 | 9.67 |
| 4.0 | 6.98 | 8.34 | 8.89 | 8.65 | 10.44 | 10.32 | 6.61 | 7.17 | 8.80 |
*The percentage of drug bound was calculated as (Db/Dt) x 100. Db was obtained as D t -D f and D f at different drug:protein ratios was obtained from the linear D f versus D t /Pt plots
Table 1: Percentage of drug bound at different drug: protein ratios.
The association constants for the binding, determined by the non-linear least squares regression program, are given in Table 2. The experimental data could be fitted into an equation for only one class of binding sites (m=1). Association constants were of the order of 104. The order of association constants was dicloxacillin> cloxacillin> flucloxacillin. The nature of drug-protein interaction could be predicted from the thermodynamic parameters for the binding. Thermodynamic parameters for the binding, determined from the temperature-dependence of the association constants, are given in Table 3. For electrostatic interactions, ΔH° has a very small negative value, nearly zero [15,22]. In the present case, high positive enthalpy changes indicate that electrostatic interactions are not present. This can also be inferred from the fact that at physiological pH, drugs as well as BSA are in predominantly anionic form. Positively charged side chain residues of protein do not appear to be involved in binding. Both ΔH° and ΔS° were found to be positive in the case of all the three drugs. The magnitude of ΔH° and ΔS° values was relatively higher for dicloxacillin and flucloxacillin as compared to cloxacillin. High positive ΔS° and ΔH° values in the case of all the three penicillins indicate that hydrophobic interactions are predominantly involved [23]. Landau et al. [24] and others [11], using microcalorimeteric technique, have also shown that for cloxacillin and dicloxacillin, the interaction is essentially hydrophobic in nature.
| Temperature (K) | Cloxacillin | Dicloxacillin | Flucloxacillin | ||||
|---|---|---|---|---|---|---|---|
| K* x 104 | n | K* x 104 | n | K* x 104 | n | ||
| 283.15 | 1.13 | 0.98 | 1.23 | 1.11 | 0.79 | 1.02 | |
| 290.15 | 1.29 | 1.11 | 2.34 | 0.98 | 0.91 | 0.99 | |
| 300.15 | 1.34 | 1.03 | 3.37 | 0.93 | 1.50 | 1.00 | |
*K and n are the association constant and the number of binding sites
Table 2: Binding parameters at ph 7.4 and different temperatures.
| Drug sample | Free energy change (?G°)* kJ mol-1 | Enthalpy change (?H°) kJ mol-1 | Entropy change (?S°) J mol-1 |
|---|---|---|---|
| Cloxacillin | -23.71 | +6.81 | +101.87 |
| Dicloxacillin | -26.01 | +41.02 | +223.91 |
| Flucloxacillin | -23.99 | +27.11 | +170.01 |
*The reported values are at 300.15 K
Table 3: Thermodynamic parameters for the binding of various isoxazolyl penicillins to serum albumin
In order to further understand the nature of interaction involved, binding was also studied in the presence of hydrophobic probe ANS. Under identical conditions, whereas ANS could quench about 70% of protein fluorescence, the drugs used could quench only 10-15% of the protein fluorescence. Thus, the percentage quenching of protein fluorescence by drugs was much less as compared to the quenching by ANS. It thus appears that the drugs and ANS do not share common site on the albumin molecule. ANS fluorescence was also measured in albumin-ANS mixture in the absence and presence of increasing amounts of drug. It was found that in each case, the presence of drug caused only a small decrease in the fluorescence of ANS. The effect was quantitatively studied by determining the percentage displacement of probe ANS (D). The results showed that up to a drug:protein ratio of 7:1, only 3-6% of drugs were displaced, which again leads to the conclusion that the drugs and ANS do not share common site on albumin.
Stern-Volmer analysis of fluorescence data is useful in the estimation of the accessibility of tryptophan residues in proteins to the drug (quencher) molecules. Fluorescence quenching data was therefore analysed by the modified Stern-Volmer equation [25],
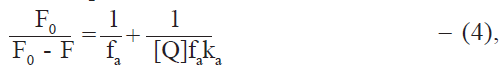
where [Q] = Dt, the total drug concentration; F0 and F are the steady state fluorescence intensities at 344 nm in the absence and presence of quencher (drug), respectively; Kq is the Stern-Volmer quenching constant; and fa is the fraction of fluorophore accessible to the quencher (Table 4). From the linear F0/(F0-F) versus 1/Dt plots, Kq and fa values were calculated. Kq values were of the order of 104 and fa values were found to lie between 0.64 and 0.72 in most cases. It thus appears that the tryptophan residues of protein are not fully accessible to drugs. This may be due to the different environments of the two tryptophan residues in albumin molecule. fa values higher than 0.5, however, suggest that both the tryptophan residues are involved. Steinhardt et al. [26] have suggested that the environment of the tryptophan residue located at position 134 of BSA has higher polarity than the tryptophan located at position 212. Since the drugs used are relatively polar in nature due to the presence of carboxyl group, it appears that tryptophan 134 is more easily accessible than tryptophan 212 in the binding of these drugs to BSA. No explanation could be found for the exceptionally low fa value (0.38) in the case of dicloxacillin at 300.15 K. The observation that the quenching constants are close to the association constants for the interaction indicate that the quenching mechanism essentially involves static quenching [27].
| Temperature (K) | Cloxacillin | Dicloxacillin | Flucloxacillin | |||
|---|---|---|---|---|---|---|
| Kq | fa | Kq | fa | Kq | fa | |
| 283.15 | 1.01 | 0.82 | 1.06 | 0.71 | 0.78 | 0.69 |
| 290.15 | 1.28 | 0.64 | 1.24 | 0.69 | 0.85 | 0.72 |
| 300.15 | 1.25 | 0.66 | 2.52 | 0.38 | 1.37 | 0.67 |
Quenching constant (Kq) and the fraction of fluorophore accessible to drug (fa) have been obtained from the modified Stern-Volmer equation (Eq. 4).
Table 4: Parameters of the stern-volmer equation.
It may thus be concluded that isoxazolyl penicillins are bound to serum albumin on a single site with moderate affinity. The binding is predominantly through hydrophobic interactions. Studies carried out in the presence of hydrophobic probe showed that the drugs and ANS do not share a common site on the albumin molecule. Stern-Volmer analysis of the fluorescence data showed that both the tryptophan residues of BSA are involved in binding, but tryptophan 134 is more easily accessible to these drugs than tryptophan 212. Moreover, static quenching mechanism appears to be involved in the case of all the drugs.
Acknowledgements
Pooja Agarwal, one of the authors, is thankful to the Council of Scientific and Industrial Research (CSIR), New Delhi, India, for financial assistance. The authors are also thankful to M/s Surya Medicare Ltd., Derabassi, India, for gift samples of the drugs.
References
- Basker, M.J., Edmondson, R.A. and Sutherland, R., J. Antimicrob.Chemother.,1982, 9, 239.
- Lacey, R., J. Antimicrob. Chemother., 1980, 6, 333.
- Mandell, G.L. and Petri, W.A. Jr., In; Hardman, J.G., Limbird, L.E. and Gilman, A.G., Eds., Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 10th Edn., McGraw-Hill, New York, 2001, 1073.
- Terasaki, T., Nouda, H. and Tsuji, A., J. Pharmacobio-Dynamics, 1992, 15, 99.
- Nouda, H., Tamai, I., Terasaki, T. and Tsuji, A., J. Pharmacobio-Dynamics, 1986, 9, s134.
- Keen, P.M., Biochem. Pharmacol., 1966, 15, 447.
- Craig,W.A. and Kunin, C.M., Ann. Rev. Med., 1976, 27, 287.
- Barbosa, S., Taboada, P., Attwood D. and Mosquera, V., Langmuir, 2003, 19, 10200.
- Barbosa, S., Taboada, P. and Mosquera, V., Langmuir, 2003, 19, 1446.
- Ruso, J.M., Attwood, D., Garcia, M., Taboada, P., Varela, L.M. andMosquera, V., Langmuir, 2001, 17, 5189.
- Markovich, M.N., Isakovich, L.G. and Klinichev, V.F., Antibiot.Med. Biotekhnol., 1986, 31, 603.
- Ullmann, U., Arzneim. Forsch-Drug Res., 1977, 27, 2136.
- He, X.M and Carter, D.C., Nature, 1992, 358, 209.
- Job, P., Ann. Chim., 1928, 9, 113.
- Rahman, M.H., Maruyama, T., Okada, T., Yamasaki, K. and Otagiri, M., Biochem. Pharmacol., 1993, 46, 1721.
- Ward, L.D., Meth. Enzymol., 1985, 117, 400.
- Weber, G. and Young, L.B., J. Biol. Chem., 1964, 239, 1415.
- Maruyama, T., Otagiri, M. and Schulman, S.G., Int. J. Pharm., 1990, 59, 137.
- Daniel, E. and Weber, G., Biochemistry, 1966, 5, 1893.
- Seedher, N., Indian J. Pharm. Sci., 1999, 62, 16.
- Silva, D., Cortez, C.M. and Louro, S.R.W., Braz. J. Med. Biol.Res., 2004, 37, 963.
- Ross, P.D. and Subramanian, S., Biochemistry, 1981, 20, 3096.
- Aki, H. and Yamamoto, M., J. Pharm. Pharmacol., 1989, 41, 674.
- Landau, M.A., Markovich, M.N. and Piruzyan, L.A., Biochim.Biophys. Acta, 1977, 493, 1.
- Lehrer, S.S., Biochemistry, 1971, 10, 3254.
- Steinhardt, J., Leidy, J.G. and Mooney, J.P., Biochemistry, 1972, 11, 1809.
- Gonzalez-Jimenez, J., Frutos, G. and Cayre, I., Biochem.Pharmacol. , 1992, 44, 824.





