- *Corresponding Author:
- Xuelian Zhao
Department of Neurosurgery, Tsinghua Changgung Hospital, Tsinghua University, Changping, Beijing 102218, China
E-mail: muruiwa@163.com
| This article was originally published in a special issue, “Exploring the Role of Biomedicine in Pharmaceutical Sciences” |
| Indian J Pharm Sci 2024:86(1) Spl Issue “86-95” |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
To construct a risk model using 760 tumor microenvironment-related gene expressions in glioblastoma patients treated with precision medicine thyrointegrin alpha v beta 3 antagonist and explore the relationships between prognosis and other clinical features and risk model is the objective of the study.Firstly, we obtained 760 tumor microenvironment-related gene expressions in the glioblastoma cells treated with 30 µM concentration of thyrointegrin alpha v beta 3 antagonist. Meanwhile, we selected genes that are significantly related to prognosis using univariable Cox regression methods. Least absolute shrinkage and selection operator analysis was applied to further select genes that are significantly related to prognosis. We weighted those selected genes with their coefficients and constructed a risk model. The risk score was calculated. We compared overall survival between low risk group and high risk group and investigated whether risk score could serve as an independent factor affecting prognosis. Furthermore, the key immune infiltrations in high and low risk groups were also deeply explored.Low risk group exhibited a better prognosis than high risk group. The multivariate Cox analysis demonstrated that risk score was an independent factor for glioblastoma prognosis. High risk group was related to a higher degree of immune infiltration.The risk model constructed by 760 tumor microenvironment-related gene expression in glioblastoma was significantly related to prognosis and this model provided several beneficial hints for the novel precision drug administration.
Keywords
Glioblastoma, tumor microenvironment, thyrointegrin alpha v beta 3 antagonist, Cox regression methods
Glioma is one of the most common primary tumors of the central nervous system[1]. Different gliomas are graded according to the World Health Organization (WHO) guidelines for classifying central nervous system tumors, WHO grades II, III and IV, where WHO grade IV diffuse glioma being synonymous with glioblastoma[2]. However, the most common diffuse glioma is glioblastoma (WHO grade IV), which accounts for 45 %-50 % of all primary intrinsic brain tumors, with the vast majority of glioblastoma arising de novo as "primary glioblastomas"[3]. Glioblastoma Multiforme (GBM) is one of the most lethal brain tumor, with an average survival rate of 35.7 % in 1 y, 4.7 % in 5 y and a median total survival period of 14.6 mo[3,4]. In 2010, based on the molecular features of GBM bulk sequencing, Wen et al. classified GBM into four subtypes. They are classical, neural, proneural and mesenchymal[5]. However, the neural type was later excluded[6]. In 2016, the WHO classification was updated to integrate molecular parameters with histology and to divide GBM into three subtypes. They are Isocitrate Dehydrogenase (IDH) wild type, IDH mutant and Not Otherwise Specified (NOS)[7]. In recent years, more treatment options have been developed for GBM, including conventional surgical resection, chemotherapy, radiotherapy, immunotherapy and electric field therapy[8]. However, the overall survival of the disorder has been reported to be <15 mo after diagnosis[9] and 5 y survival rates ranged from only 0.05 % to 4.7 %[10].
It is not only tumor cells but also surrounding normal cells that play the key role in the progress of tumor. These normal cells constitute the Tumor Microenvironment (TME). The TME comprises different cellular components. The first type is endothelial cells, which play a critical role in tumorigenesis and in protecting tumor cells from the immune system. Tumor angiogenesis usually branches outward from pre-existing vessels or arises from endothelial precursors[11]. In this way, these cells provide nutrients for tumor cell proliferation and growth. The second major component is immune cells. These include granulocytes, lymphocytes and macrophages. The most prominent immune cell type in the TME is the macrophage. Macrophages have multiple functions associated with cancer development and progression; they promote tumor cell escape into the circulation and suppress anti-tumor immune mechanisms and responses[12]. The last cell type in the TME is the fibroblast. Fibroblasts are responsible for the migration of cancer cells from the primary tumor site into the blood stream for systemic metastasis[13,14].
Thyrointegrin alpha v beta 3 (αvβ3) antagonist, considered as a bifunctional version of the thyroid hormone metabolite Tetraiodothyroacetic acid (Tetrac) conjugated to Polyethylene Glycol (PEG) MW 4000, was originally initiated as an innovative new precision medicine for GBM. It has been demonstrated with an excellent efficacy in GBM mouse model. However, the accurate function of thyrointegrin αvβ3 antagonist in human is still poorly understood. Meanwhile, some works have suggested the potential connections between thyrointegrin αvβ3 antagonist and TME.
In this study, we selected genes that have an impact on prognosis from the GBM tumor cells treated with thyrointegrin αvβ3 antagonist of 760 TME related genes. Then we constructed a risk model related with those selected genes. Using this risk model, we explored a close correlation between the TME of GBM and thyrointegrin αvβ3 antagonist.
Materials and Methods
Data source:
In this study, we used two independent data sources. The glioblastoma patients from Gene Expression Omnibus (GEO) database (numbering GSE183772) were considered as a constructing database. It includes GBM cells treated with thyrointegrin αvβ3 antagonist. At the same time, the glioblastoma patients from Chinese Glioma Genome Atlas (CGGA) database were treated as a testing database.
Least Absolute Shrinkage and Selection Operator (LASSO) Cox regression analysis:
A single-factor Cox regression analysis for glioblastoma prognosis was developed to analyze the expression values of 760 TME-related genes in glioblastoma samples, with a threshold of p<0.01. Essentially, the complete list of genes was obtained from 10 published studies providing transcriptomic signatures for multiple immune and stromal cell populations[15-24]. Then, to further infer the TME genes related to glioblastoma prognosis, glmnet package of R language was used for LASSO Cox regression analysis. The following formula was established to calculate the risk score for each individual glioblastoma sample:
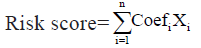
In this formula, Coefi represents the risk coefficient of each factor estimated by the LASSO-Cox model, while Xi indicates the expression activity of each TME gene. Based on the median of the calculated risk scores, the glioblastoma patients of the construction database and the test database could be divided into high risk group and low risk group.
Gene set enrichment analysis:
In order to explore different pathways between high risk group and low risk group, gene set enrichment analysis was conducted[25]. The top 10 terms of the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Molecular Signatures Database (MSigDB) hallmark analyzes were displayed. KEGG pathways with significant enrichment results were displayed based on Normalized Enrichment Score (NES), gene ratio and p value. Gene sets with |NES|>1, Nominal (NOM) p<0.05 and False Discovery Rate (FDR) q<0.25 were considered enrichment significant.
Somatic mutation analysis:
The Cancer Genome Atlas (TCGA)-GBM contains genome sequencing data, among them we downloaded simple nucleotide variation data’s from the website (https://portal.gdc.cancer.gov/) to compare the mutation frequency of each gene of the high and low risk group. The somatic mutation landscape and the mutation frequency of each gene was realized by the maftool R package[26].
Immune infiltration analysis:
To study the immune infiltration in glioblastoma, we used three algorithms: ESTIMATE[27], Cell-type Identification by Estimating Relative Subsets of RNA Transcripts (CIBERSORT)[28], single-sample Gene Set Enrichment Analysis (ssGSEA)[29]. CIBERSORT is a linear support vector regression based tool for deconvolution of human leukocyte subtype expression matrix from bulk tissue gene expression profiles. Input was a 547 signature gene matrix containing 22 functionally defined human immune subsets (LM22). The data were uploaded to the CIBERSORT web portal (http://cibersort.stanford.edu/) and iterated 1000 times to obtain results. ESTIMATE is a tool that uses gene expression signatures to infer the proportion of stromal cells and the proportion of immune cells in a tumor sample. The score derived from ssGSEA reflects the degree to which the input immune gene signature is coordinately up-regulated or downregulated within a sample.
Drug sensitivity analysis:
National Cancer Institute (NCI)-60 compound activity data and Ribonucleic Acid sequencing (RNA-seq) expression profiles were downloaded from CellMiner™ to analyze drug sensitivity of selected TME related genes in TCGA-GBM. Food and Drug Administration (FDA)-approved or investigational drugs were selected to analyze[30].
Immunophenoscore (IPS) analysis:
Based on four major gene categories that determine immunogenicity, an IPS consisting of Major Histocompatibility Complex (MHC), Effector Cells (ECs), immune Checkpoints (CPs) and Immunosuppressive Cells (ISCs) can be generated using machine learning methods. Immunophenotype scores, ranging from 0 to 10, were calculated using the expression levels of representative genes or the immune manifestation of gene sets. The IPS of glioblastoma patients were obtained from The Cancer Immunome Atlas (TCIA) database (https://tcia.at/home).
Statistical analysis:
All statistical methods were executed on R software (v4.2.3). To compare two or more continuous variables, the unpaired Student’s t-test was used for data that followed a normal distribution and the Wilcoxon or Kruskal-Wallis test was applied for non-normally distributed data. Based on the Kaplan-Meier method, the survival R package was utilized to generate survival curves and the log-rank test was established to determine the significance of differences. The univariate and multivariate Cox regression model was used to determine independent prognostic factors by using the survminer package and the results was visualized by ggforest package. The prognostic model of glioblastoma was constructed using the nomogram function of the regression modeling strategies (rms) R package. Two-sided p<0.05 was considered as statistically significant.
Results and Discussion
TME-related gene selection and the construction of risk score was shown in fig. 1A-fig. 1E. Firstly, as a construction database we downloaded GEO database. We focused on the TME genetic profiling for glioblastoma patients treated with thyrointegrin αvβ3 antagonist. The expression of 760 TME related genes in the database was obtained. The selection was based on a complete list of 760 TME genes. We used the expressions of these genes to establish a univariate cox regression to screen for genes that have an impact on prognosis after medicine treatment. 46 genes were selected as p value<0.01. Among the selected genes, genes with Hazard Ratio (HR)>1 are unfavorable for prognosis (risk genes), while genes with HR<1 are favorable for prognosis (protective genes). To this end, 29 of the 46 genes were considered as protective genes, while the remaining 17 genes were considered as risk genes. To further select genes significantly associated with prognosis, we conducted LASSO regression analysis. The horizontal axis is log (lambda) and the vertical axis is the partial likelihood deviation value respectively. The lambda value corresponding to the smallest value is the best. As shown in fig. 1A, the optimal number of significant impact on prognosis of TME genes was determined as 17, whereas the lambda value is the smallest. Therefore, the forest plot of the top 17 genes with the smallest p-value among the 46 genes is shown in fig. 1B. A risk score model for predicting the survival of glioblastoma patients was then generated by weighing the expression values of 17 TME genes with the regression coefficients of the LASSO Cox regression model. The risk score of each patient was calibrated individually. Kaplan-Meier survival curve of TCGA-GBM dataset was shown in fig. 1C. The horizontal axis indicates time, while the vertical axis indicates survival rate. The p-value is calculated based on log-rank test and different color means different group. The median value of risk score was used to classify glioblastoma patients into high risk and low risk groups. Survival analysis was conducted on the prognosis of patients in the high risk and low risk groups, and compared to the low risk group, the high risk group had a poorer prognosis (fig. 1C). In addition, it could be shown that the Area Under the Curve (AUC) of 1 y, 3 y and 5 y survival period of glioblastoma patients were 0.790, 0.809 and 0.919, respectively, conducted from the time-dependent Receiver Operating Characteristic (ROC) curve. The horizontal axis means the false positive rate, while the vertical axis means the true positive rate respectively. The accuracy of the prediction is evaluated by the AUC value (fig. 1D). These results indicated that the risk model could accurately predict the prognosis of glioblastoma patients with thyrointegrin αvβ3 antagonist treatment. The distribution-risks of the risk scores, survival time, survival status and the expression patterns of 18 genes in constructing database and testing database are displayed in fig. 1E. To compare risk score between different ages, we divided glioblastoma patients in construction database into two age groups: Age above 60 group and age under 60 group. We also compared risk score between different genders. Risk score showed no significant differences between different age and different gender groups.

Fig. 1: TME-related gene selection and the construction of risk score.
Note: (A) The graph determining the tuning parameter lambda value in the LASSO regression model; (B) Forest plot of top 17 TME-related
genes with smallest p value; (C) Kaplan-Meier survival curve of TCGA-GBM dataset,  (D) The
time-dependent ROC curve,
(D) The
time-dependent ROC curve,  and (E) Distribution of the expression profiles, survival status and risk score
of TCGA-GBM database,
and (E) Distribution of the expression profiles, survival status and risk score
of TCGA-GBM database,  Survival time,
Survival time,  risk group.
risk group.
To validate this risk model, we utilized another testing database CGGA. This model was established as a testing database, calculating risk score of each sample. Then we divide glioblastoma patients in testing database into high risk group and low risk group using median value of risk score. The overall survivals between high risk group and low risk group were compared, suggesting that high-risk group also had a lower survival than low risk group. The distribution-risks of the risk scores, survival time, survival status and the expression patterns of 17 genes in testing database are also displayed. Then we compared risk score in different clinical characteristics such as age (age above 60 y or age under 60 y), gender, received thyrointegrin αvβ3 antagonist or not, received radiotherapy or not and IDH mutation status (IDH mutation or wild type) in testing database. As a result, there was no significant difference in risk score between genders. The risk score was significantly higher than the IDH mutation group which suggested that IDH mutation might be a good prognostic marker for glioblastoma. Meanwhile, patients older than 60 y old exhibited a higher risk score than patients younger than 60 y old. There was no significant difference in risk score between receiving chemotherapy and radiation therapy.
Pathway analysis and somatic mutation analysis was shown in fig. 2A-fig. 2E. In order to explore different biological functions between high risk group and low risk group in thyrointegrin αvβ3 antagonist treated GBM, differential analysis and subsequently GSEA analysis were conducted. The limma package was used to conduct differential analysis. To visualize the results of differential analysis between high risk group and low risk group, volcano plot was plotted. Both p value<0.05 and log2 Fold Change (FC)>1 means up-regulated genes and both p value<0.05 and log2FC<-1 means down-regulated genes (fig. 2A). Then GSEA analysis was conducted. GSEA results showed that tumors in the high risk score group had gene expressions significantly enriched in pathways related to immunity such as adaptive immune response based on antigen binding, complement activation, humoral immune response, leukocyte migration, regulation of humoral immune response (fig. 2B), whereas the low risk score tumors had gene expressions enriched in chromosomal region, mitochondrial gene expression, non-coding RNA (ncRNA) metabolic process, ncRNA processing, ribosome biogenesis (fig. 2C). Meanwhile, in testing database, GSEA was also conducted. Tumors in the high risk score group also exhibited gene expressions significantly enriched in pathways related to immunity. While tumors in the low risk score group has gene expressions mainly enriched in synapse-related pathway.

Fig. 2: Pathway analysis and somatic mutation analysis.
Note: (A) The volcano plot of differential expressed genes between high risk group and low risk group,  Down-regulated
genes;
Down-regulated
genes;  (B, C) GSEA pathway analysis of differential expressed genes between high risk group and low risk group. The top five
pathways are displayed, (B) Pathways enriched in high risk group; (C) Pathways enriched in low risk group; (D, E) Comparisons of the mutation
landscape in the TCGA-GBM database between groups with high and low risk score, (D) High risk group (E) Low risk group.
(B, C) GSEA pathway analysis of differential expressed genes between high risk group and low risk group. The top five
pathways are displayed, (B) Pathways enriched in high risk group; (C) Pathways enriched in low risk group; (D, E) Comparisons of the mutation
landscape in the TCGA-GBM database between groups with high and low risk score, (D) High risk group (E) Low risk group.
We also examined the mutation landscapes between patients with low and high risk scores. The top 5 genes with mutation frequency in the high risk group were Phosphatase and Tensin homolog (PTEN), Titin (TTN), Epidermal Growth Factor Receptor (EGFR), Tumor Protein 53 (TP53) and LDL Receptor Related Protein 2 (LRP2) (fig. 2D). Meanwhile, the top 5 genes with mutation frequency in the low risk group were TP53, EGFR, PTEN, TTN and Mucin 16 (MUC16) (fig. 2E). Since IDH1 mutation is considered as one of the factors related to the prognosis of glioblastoma treated with thyrointegrin αvβ3 antagonist[31], we compared the frequency of IDH1 mutation in the high and low risk groups. The mutation frequency of IDH1 in the low risk group was higher than that in the high-risk group, which indicated glioblastoma with IDH1 mutation had a better prognosis.
Risk score is an independent prognostic factor for glioblastoma as shown in fig. 3A-fig. 3E. To determine whether risk score can be an independent factor affecting prognosis, the age, gender and risk score of each individual glioblastoma patients were all subjected in the multivariate Cox regression analysis. As a result, we found a dramatic connection between risk score and the prognosis of glioblastoma, where the higher the risk score, the worse the prognosis (fig. 3A). We constructed a nomogram plot to predict the survival of Neuroblastoma (NB) patients to make the new model (risk score, gender and age) more applicable to clinical practice. A comprehensive score was assigned to each patient. This score was calculated by adding the corresponding scores for each variable in the nomogram plot (fig. 3B). 1 y and 2 y calibration curves were used to determine the accuracy and clinical utility of the comprehensive score and the new model (fig. 3C). To investigate the prognostic value of the risk score established by 17 potential TME-related genes, we further regrouped patients from the constructing database and performed Kaplan-Meier survival analysis. Patients were divided into group A (<60 y old) and group B (≥60 y old). Patients were defined as high risk or low risk based on the risk score established by 17 potential TME-related genes. Regardless of age, patients in the high risk group had a significantly lower overall survival rate than those in the low risk group (fig. 3D and fig. 3E). These results concluded that the risk score built by primary TME genes was an independently accurate indicator for predicting the prognosis of glioblastoma patients treated with thyrointegrin αvβ3 antagonist.

Fig. 3: Risk score as an independent prognostic factor for glioblastoma.
Note: (A) Multivariate cox regression model of risk score and clinical characteristics of glioblastoma patients in TCGA-GBM; (B) Nomogram plot
for predicting the probability of patient mortality at 1 y or 2 y overall survival; (C) Calibration curve of nomogram for predicting the probability
of overall survival at  (D, E) After regrouping according to age 60, Kaplan-Meier survival curve of two groups, (D)
Kaplan-Meier survival curve of age≥60 group and (E) Kaplan-Meier survival curve of age<60 group,
(D, E) After regrouping according to age 60, Kaplan-Meier survival curve of two groups, (D)
Kaplan-Meier survival curve of age≥60 group and (E) Kaplan-Meier survival curve of age<60 group,  score.
score.
Risk score is related to immune infiltration which was illustrated in fig. 4A-fig. 4G. The infiltration of immune cells in tumor cells plays a crucial role in glioblastoma progression. To explore correlation between immune infiltration and risk score, ESTIMATE, CIBERSORT and ssGSEA analysis were conducted. Firstly, we calculated stromal score, immune score, estimate score and tumor purity of each tumor sample using ESTIMATE algorithm and we constructed correlation between those scores and risk score. As for constructing database, results demonstrated that there was significant positive correlation between stromal score, immune score, estimate score and risk score. Meanwhile, there was significant negative correlation between tumor purity and risk score (fig. 4A-fig. 4D). There was also a similar correlation in the testing database. We divided patients into high risk and low risk groups in constructing database and testing database. We also estimated the immune cell infiltration both in high and low risk score groups, respectively, by the CIBERSORT and ssGSEA algorithm. Using CIBERSORT in constructing database, we found that Cluster of Differentiation (CD) 8 T cells, activated CD4 memory T cells and activated mast cells were highly infiltrated in high risk group. Resting mast cells and activated Natural Killer (NK) cells were highly infiltrated in low risk group. In testing database, activated NK cells were highly infiltrated in low risk group. M0 macrophages and neutrophils were highly infiltrated in high risk group (fig. 4E). In constructing database ssGSEA analysis showed that except memory B cells, all immune cells are highly infiltrated in high risk group (fig. 4F). In testing database, type 2 T helper cells are highly infiltrated in low risk group and memory B cells, eosinophils, CD4 effector memory T cells, activated CD4 T cells and CD56 bright NK cells showed no significant between low risk group and high risk group. Other immune cells were highly infiltrated in high risk group. Heatmap of ssGSEA in constructing database indicated that there was a significant difference in immune infiltration between the high risk and low risk group (fig. 4G).

Fig. 4: Relation of risk score with immune infiltration.
Note: Scatter plot shows correlation between (A) Stromal score; (B) Immune score; (C) Estimate score; (D) Tumor purity and risk score; (E) Boxplot
of 22 immune cells fraction between  using CIBERSORT; (F) Boxplot of immune cells infiltration
comparison between
using CIBERSORT; (F) Boxplot of immune cells infiltration
comparison between  using ssGSEA; (G) Heatmap of 28 immune cells fraction in high risk group
and low risk group,
using ssGSEA; (G) Heatmap of 28 immune cells fraction in high risk group
and low risk group,  . The p values were calculated by Wilcoxon rank-sum test. The asterisks represented
the statistical p value (*p<0.05; **p<0.01; ***p<0.001 and ****p<0.0001).
. The p values were calculated by Wilcoxon rank-sum test. The asterisks represented
the statistical p value (*p<0.05; **p<0.01; ***p<0.001 and ****p<0.0001).
Drug sensitivity analysis and immune reaction prediction was shown in fig. 5A-fig. 5E. Using the CellMiner database, we further investigated the potential correlation between drug sensitivity of thyrointegrin αvβ3 antagonist and the expression of 17 TME-related genes. The expression of 17 TME-related genes in cell lines for studying drug sensitivity was obtained. The correlation between drug sensitivity and the expression of 17 inflammation-related genes was obtained (p<0.05 was set as a threshold), and top 16 correlation was shown in fig. 5A. Then, we used IPS to predict immune response. The IPS score of glioblastoma patients were obtained from TCIA. We divided patients into four types (i.e., IPS-Cytotoxic T-Lymphocyte-Associated protein-4 (CTLA4)-negative (neg)-Programmed Cell Death protein 1 (PD-1)-neg, IPS-CTLA4-neg-PD1-positive (pos), IPS-CTLA4-pos-PD1-neg and IPS-CTLA4-pos-PD1-pos) and comparing their risk score. It is obvious that patients with low risk score expressed greater IPS values when only receiving CTLA4 treatment and not receiving CTLA4 and PD1 treatment (fig. 5B-fig. 5E).

Fig. 5: Drug sensitivity analysis and immune reaction prediction
Note: (A) Scatterplot shows correlation between drug sensitivity and the expression of 17 inflammation-related genes in cell lines conducted by
CellMiner; Comparison of IPS score between high risk score and low risk score under different immune therapy treatment; (B) CTLA4-pos-PD1-
pos treatment; (C) CTLA4-pos-PD1-neg treatment; (D) CTLA4-neg-PD1-pos treatment and (E) CTLA4-neg-PD1-neg treatment. The p values were
calculated by Wilcoxon rank-sum test,  .
.
Glioma is a common central nervous system tumor that can be classified into four grades according to the WHO classification criteria, with the IV grade being called glioblastoma[2]. Glioblastoma is the most common brain tumor with a poor prognosis. TME is a complex ecosystem composed of malignant, stromal and immune cells[32]. Cells in TME can promote tumor growth and proliferation. TME has a significant impact on the prognosis of glioblastoma. In this study, we constructed a risk model using the expression profile of TME related genes that have a significant impact on prognosis.
Firstly, using constructing database TCGA-GBM, we obtained the expression profiles of 760 TME related genes in glioblastoma. Then we used univariate Cox regression to select 46 genes with p<0.01. These 46 genes were further featured screened using LASSO regression analysis and 17 genes were selected. Among them 9 genes (Lysyl Oxidase Like 1 (LOXL1), Adipocyte Enhancer-Binding Protein 1 (AEBP1), Insulin-like Growth Factor Binding Protein 6 (IGFBP6), Retinoic Acid Receptor Responder 2 (RARRES2), EGF containing Fibulin Extracellular Matrix Protein 2 (EFEMP2), Integrin subunit Alpha 3 (ITGA3), LOXL4, Homeobox B2 (HOXB2) and Canopy FGF Signaling Regulator 4 (CNPY4)) are unfavorable for overall survival. Meanwhile, 8 genes (FRY Microtubule Binding Protein (FRY), FRAT Regulator of WNT signaling pathway 2 (FRAT2), WD Repeat Domain 43 (WDR43), Radixin (RDX), Splicing Factor 3A Subunit 3 (SF3A3), Fermitin family homolog 2 (FERMT2), Adenylosuccinate Lyase (ADSL) and ADP Ribosylation FACTOR Like GTPase 2 Binding Protein (ARL2BP)) are favorable for overall survival. We weighed those 17 gene expression with their coefficients calculated by LASSO to construct a risk score model. Median value of risk score was used to divide patients into high risk group and low risk group. We also in-depth explored those 17 genes and their corresponding coefficients to construct a risk score model in testing database. The prognosis of glioblastoma patients in the low risk group was significantly better than that in the high risk group. Compared to the low risk group, the differentially expressed genes in the high risk group were mainly enriched related to immunity-related pathway suggesting that immunity plays an intriguing role in the occurrence and development of glioblastoma. The frequency of IDH1 mutations was higher in low risk glioblastoma patients than in high risk ones, indicating IDH1 mutation is a positive factor affecting prognosis. We proved that risk score is an independent factor for glioblastoma using forest plot and nomogram. As for immune infiltration, ESTIMATE analysis showed that risk score had a positive correlation with stromal feature and immune feature and risk score has negative correlation with tumor purity. ssGSEA confirmed that high risk group had a higher immune infiltration than low risk group. Then we conducted drug sensitivity analysis and constructed correlation with 17 selected genes expression in cell lines.
Finally, we found correlation between immune therapy reaction and risk score. In low risk score group, IPS score were higher than that in high risk group. All in all, we constructed a risk model using selected TME-related genes and we found correlations between risk score and prognosis.
Conflict of interests:
The authors declared no conflict of interest.
References
- Weller M, Wick W, Aldape K, Brada M, Berger M, Pfister SM, et al. Glioma. Nat Rev Dis Primers 2015;1(1):1-8.
[Crossref] [Google scholar] [PubMed]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007;114(2):97-109.
[Crossref] [Google scholar] [PubMed]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352(10):987-96.
[Crossref] [Google scholar] [PubMed]
- Perry JR, Laperriere N, O’Callaghan CJ, Brandes AA, Menten J, Phillips C, et al. Short-course radiation plus temozolomide in elderly patients with glioblastoma. N Engl J Med 2017;376(11):1027-37.
[Crossref] [Google scholar] [PubMed]
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med 2008;359(5):492-507.
[Crossref] [Google scholar] [PubMed]
- Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer cell 2017;32(1):42-56.
[Crossref] [Google scholar] [PubMed]
- Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol 2016;131(6):803-20.
[Crossref] [Google scholar] [PubMed]
- Alifieris C, Trafalis DT. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol Ther 2015;152:63-82.
[Crossref] [Google scholar] [PubMed]
- Rajaratnam V, Islam MM, Yang M, Slaby R, Ramirez HM, Mirza SP. Glioblastoma: Pathogenesis and current status of chemotherapy and other novel treatments. Cancers 2020;12(4):937.
[Crossref] [Google scholar] [PubMed]
- Ostrom QT, Bauchet L, Davis FG, Deltour I, Fisher JL, Langer CE, et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol 2014;16(7):896-913.
[Crossref] [Google scholar] [PubMed]
- Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell 2011;144(5):646-74.
[Crossref] [Google scholar] [PubMed]
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature 2008;454(7203):436-44.
[Crossref] [Google scholar] [PubMed]
- LeBleu V. Imaging the tumor microenvironment. Cancer J 2015;21(3):174-8.
[Crossref] [Google scholar] [PubMed]
- Korneev KV, Atretkhany KS, Drutskaya MS, Grivennikov SI, Kuprash DV, Nedospasov SA. TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis. Cytokine 2017;89:127-35.
[Crossref] [Google scholar] [PubMed]
- Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 2016;17(1):1-20.
[Crossref] [Google scholar] [PubMed]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013;39(4):782-95.
[Crossref] [Google scholar] [PubMed]
- Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep 2017;18(1):248-62.
[Crossref] [Google scholar] [PubMed]
- Abbas AR, Wolslegel K, Seshasayee D, Modrusan Z, Clark HF. Deconvolution of blood microarray data identifies cellular activation patterns in systemic lupus erythematosus. PLoS One 2009;4(7):e6098.
[Crossref] [Google scholar] [PubMed]
- Chifman J, Pullikuth A, Chou JW, Bedognetti D, Miller LD. Conservation of immune gene signatures in solid tumors and prognostic implications. BMC Cancer 2016;16(1):1-7.
[Crossref] [Google scholar] [PubMed]
- Clancy T, Hovig E. Profiling networks of distinct immune-cells in tumors. BMC Bioinformatics 2016;17(1):1-5.
[Crossref] [Google scholar] [PubMed]
- Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol 2016;17(1):1-6.
[Crossref] [Google scholar] [PubMed]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12(5):453-7.
[Crossref] [Google scholar] [PubMed]
- Racle J, de Jonge K, Baumgaertner P, Speiser DE, Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife 2017;6:e26476.
[Crossref] [Google scholar] [PubMed]
- Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160(1):48-61.
[Crossref] [Google scholar] [PubMed]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545-50.
[Crossref] [Google scholar] [PubMed]
- Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res 2018;28(11):1747-56.
[Crossref] [Google scholar] [PubMed]
- Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4(1):2612.
[Crossref] [Google scholar] [PubMed]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12(5):453-7.
[Crossref] [Google scholar] [PubMed]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462(7269):108-12.
[Crossref] [Google scholar] [PubMed]
- Reinhold WC, Sunshine M, Varma S, Doroshow JH, Pommier Y. Using CellMiner 1.6 for systems pharmacology and genomic analysis of the NCI-60. Clin Cancer Res 2015;21(17):3841-52.
[Crossref] [Google scholar] [PubMed]
- Yao Q, Cai G, Yu Q, Shen J, Gu Z, Chen J, et al. IDH1 mutation diminishes aggressive phenotype in glioma stem cells. Int J Oncol 2018;52(1):270-8.
[Crossref] [Google scholar] [PubMed]
- Anderson NM, Simon MC. The tumor microenvironment. Curr Biol 2020;30(16):921-5.
[Crossref] [Google scholar] [PubMed]





