- *Corresponding Author:
- N. B. Patel
Department of Chemistry, M. B. Patel Science College, Charotar Education Society, Anand 388001, India
E-mail: drnavinbpatel@gmail.com
| Date of Received | 19 May 2021 |
| Date of Revision | 21 June 2023 |
| Date of Acceptance | 27 July 2023 |
| Indian J Pharm Sci 2023;85(4):1033-1044 |
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms
Abstract
1,2,4-triazole and Schiff bases are active against antimicrobial and antitubercular activities. Isoniazid itself reacts as antitubercular drug. The two dimensional quantitative structure activity relationship study was used to predicts the antitubercular activity of the synthetic derivatives. Two dimensional quantitative structure activity relationship generated model using partial least squares regression method which predict the statistically significant r2=0.8940, q2=0.8197, pred_r2=0.6675 and F test=50.6234. Two dimensional quantitative structure activity relationship generated equation of pMICs is denoted the antitubercular activity correlated with thermodynamic descriptor XAMostHydrophilic. Pharmacokinetic properties absorption, distribution, metabolism, excretion were also predicted which useful for design the derivatives. A designed derivatives of 1-(3-(substitutidene)amino)-5-(2,3,4,5-tetrafluoro-6-substitutedphenyl)-4H-1,2,4-triazol-4- yl)-3-(pyridin-4-yl)urea (B1-B10) were synthesized and spectrally evicted from fourier-transform infrared spectroscopy, 1H Nuclear magnetic resonance, 13C Nuclear magnetic resonance and Mass spectra as well as biologically evaluated against antitubercular and antimicrobial activities. From the biologically evaluated derivatives, compounds B2, B4, B7, B8, B9 and B10 were found to be active against the different antimicrobial species. Where as compound B2 also active against antitubercular species.
Keywords
Two dimensional quantitative structure activity relationship, partial least squares regression, 1,2,4-triazole, antimicrobial activity, antitubercular activity
From the last few decades, 1,2,4-triazole fused derivatives and itself it have gained high importance in the pharmaceutical industry as antimicrobial, antituberculer and antimalarial agents. Many antimicrobial drugs are available those having 1,2,4-triazole. Several significant biological activities, such as antimicrobial[1,2], antituberculosis[3], antimalarial[4], anticancer[5] and antioxidant[6] are described by 1,2,4-triazole. A number of routes are available for synthesis of 1,2,4-triazole[7-10]. From which we are synthesized 1,2,4-triazole directly from acid. Schif base is formation of carbon-nitrogen double bond which play wider role in synthetic organic chemistry. It is obtained from aldehydes and amines Schiff base creates attention for organic chemists because of its vital role in pharmaceutical chemistry. It gives significant activities like anticancer[11], antitumor[12], antituberculosis[13], antimicrobial[14], anticonvulsant[15]. Infectious deseases tuberculosis is an infectious disease which caused by Mycobacterium tuberculosis (M. tuberculosis)[16]. Today, tuberculosis is situted in the top five deases of global mortality. Approximately estimation is that arround 1 billion peoples are infected between years 2002 to 2018, around 150 million peoples are got sick and around 36 million people are died because of tuberculosis if new disease prevention and treatment measurements are not developed[17]. Isoniazid is one of the most effective antituberculosis drugs used for tuberculosis treatment.
Some few decades, Two Dimensional Quantitative Structure Activity Relationship (2D-QSAR) are being applied in many areas of drug design. Accepted technique, 2D-QSAR is carried out to predicted the pharmacological activities which is a mathematical tool used to evaluate the toxicity of a compounds from its physiochemical properties of molecular structures. Our interest for synthesis of 1,2,4-triazole, isoniazid, Schiff base and flourine containing derivatives. We are referred literature review on 2D-QSAR study of biologically active 1,2,4-triazole, isoniazid, Schiff base and flourine derivatives[18-20]. 2D-QSAR were determined by VLifeMDS software and Partial Least Squares Regression (PLSR) method. 2D-QSAR predicted value of activity which are compared with actual biological activity. Swiss Absorption, Distribution, Metabolism, Excretion (ADME) tool is used for prediction of ADME properties.
My aim is to synthesized antimicrobial and antitubercular active agents. Our interest to developed 1,2,4-triazole scaffold as a novel bio-conjugates which are exhibited pharmacological properties as an antimicrobial[21] and an antituberculer[22] activities. For this purpose, we had decided to synthesized 1,2,4-triazole based conjugates which containing Schiff base and isoniazid in its final structure.
Materials and Methods
General materials:
Analytical grade chemicals were used in synthesis. Fisher-Johns Melting Point apparatus was used to determined Melting points of synthesized compounds. Compounds purities were checked by Thin Layer Chromatography (TLC) on silica gel plates with visualization by Ultraviolet-light and iodine chamber. Fourier-Transform Infrared Spectroscopy (FTIR) spectra were analyzed by model FTIR 8400S and frequency obtained in cm-1 unit. 1H Nuclear Magnetic Resonance (NMR) and 13C Nuclear Magnetic Resonance spectra were recorded at 400 MHz on Bruker Avance II spectrometer instruments in Dimethylsulfoxide (DMSO)-d6. Chemical shifts were recorded in parts per million downfield from tetramethylsilane. Mass spectra were recorded on Liquid chromatography coupled to Mass Spectrometry (LC-MS). Structures and nomenclatures of the compounds are generated on Perkin Elmer ChemBioOffice Ultra 14.0.0.117 software. SwissADME online tool is used for prediction of absorption, distribution, metabolism, and excretion (ADME) properties.
General methods:
General procedure for synthesis of (E)-N- (5-(2,3,4,5-tetrafluoro-6-substitutedphenyl)- 1,3,4-oxadiazol-2-yl)substitutedimine (A1-A10): An equimolar mixture of 2,3,4,5-tetrafluoro-6- substitutedbenzoic acid (0.02 mol), semicarbazide (0.02 mol) and substituted aldehyde (0.02 mol) were dissolved in methanol, which succeded by the addition of acetic acid (0.02 mol) and heat up to 90° around 30 min. Than, the addition of anhydrous zinc chloride (0.02 mol), reaction mass was heated around 5 h. The TLC was checked after each 1.5 h using water:methanol (3:7) mobile phase. When completed the reaction, cooled the reaction mass and pour over crushed ice. Obtained precipitates washed with water, followed by distilled water and crystallized from methanol to make a pure product (A). All the derivatives (A1-A10) are synthesized by this method (Table 1).
| Code | Structure | R1 | R2 | % yield | MP(°) | M W gm/mol | Rf | M.F. |
|---|---|---|---|---|---|---|---|---|
| A1 | 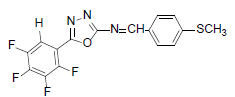 |
H | 4-Methylthiophenyl | 73 | 179 | 367.32 | 0.5 | C16H9ON3SF |
| A2 | 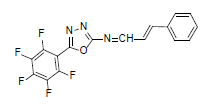 |
F | Styrene | 77 | 185 | 365.26 | 0.5 | C17H8ON3F5 |
| A3 | 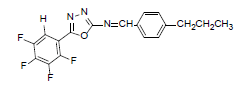 |
H | 4-Propylphenyl | 74 | 194 | 363.31 | 0.6 | C18H13ON3F4 |
| A4 | 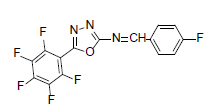 |
F | 4-Fluorophenyl | 71 | 217 | 357.21 | 0.5 | C15H5ON3F6 |
| A5 | 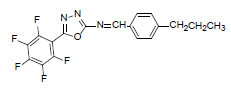 |
F | 4-Propylphenyl | 68 | 193 | 381.3 | 0.6 | C18H12ON3F5 |
| A6 | 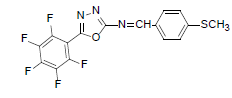 |
F | 4-Methylthiophenyl | 69 | 157 | 385.31 | 0.5 | C16H8ON3SF5 |
| A7 | 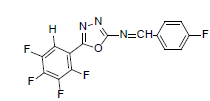 |
H | 4-Fluorophenyl | 74 | 176 | 339.22 | 0.5 | C15H6ON3F5 |
| A8 | 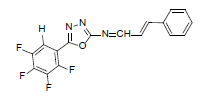 |
H | Styrene | 75 | 181 | 347.27 | 0.5 | C17H9ON3F4 |
| A9 | 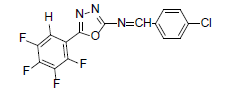 |
H | 4-Chlorophenyl | 79 | 167 | 355.67 | 0.4 | C15H6ON3F4Cl |
| A10 | 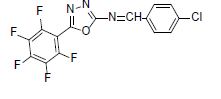 |
F | 4-Chlorophenyl | 72 | 197 | 373.66 | 0.5 | C15H6ON3F4Cl |
Table 1: Physical Data Of Compounds (A1-A10)
General procedure for the synthesis of (E)-N-(3- (substitutidene) amino)-5-(2,3,4,5-tetrafluoro- 6-Substitutedphenyl)-4H-1,2,4-triazol-4-yl) isonicotinamide (B1-B10): An equimolar mixture of compound (A) (0.02 mol), isoniazid (0.02 mol) and substituted aldehyde (0.02 mol) were dissolved in methanol in a round bottom flask, then add KOH (0.02 mol) and heat up to 90° for 4-5 h. When completed the reaction cooled the reaction mass and pour over ice cold water. Obtained precipitates washed with water, followed by distilled water and crystallized from methanol to make pure product (B). All the derivatives (B1-B10) are synthesized by this method (Table 2).
| Code | Structure | R1 | R2 | % yield | MP(°) | M. W. gm/mol | Rf | M.F. |
|---|---|---|---|---|---|---|---|---|
| B1 | 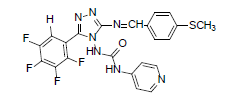 |
H | 4-Methylthiophenyl | 70 | 209 | 501.46 | 0.68 | C22H15ON7SF4 |
| B2 | 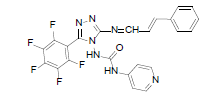 |
F | Styrene | 76 | 226 | 499.4 | 0.63 | C23H14ON7F5 |
| B3 | 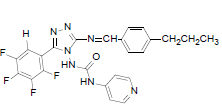 |
H | 4-Propylphenyl | 72 | 218 | 497.45 | 0.65 | C24H19ON7F4 |
| B4 | 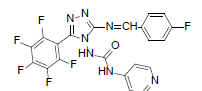 |
F | 4-Fluorophenyl | 70 | 254 | 491.35 | 0.57 | C21H11ON7F6 |
| B5 | 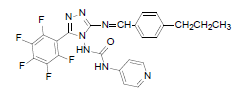 |
F | 4-Propylphenyl | 67 | 230 | 515.44 | 0.76 | C24H18ON7F5 |
| B6 | 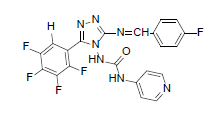 |
F | 4-Methylthiophenyl | 69 | 190 | 519.45 | 0.63 | C22H14ON7SF5 |
| B7 | |
H | 4-Fluorophenyl | 75 | 207 | 473.36 | 0.58 | C21H12ON7F5 |
| B8 | 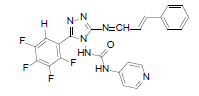 |
H | Styrene | 73 | 201 | 481.4 | 0.69 | C23H15ON7F4 |
| B9 | 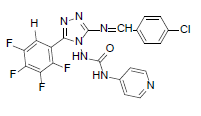 |
H | 4-Chlorophenyl | 80 | 196 | 489.81 | 0.71 | C21H12ON7F4Cl |
| B10 | 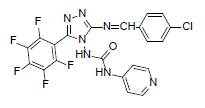 |
F | 4-Chlorophenyl | 71 | 230 | 507.8 | 0.77 | C21H11ON7F5Cl |
Table 2: Physical Data Of Compounds (B1-B10)
Spectral data of compounds:
1 - ( 4 - M e t h y l t h i o ) p h e n y l ) - N - ( 5 - ( 2 , 3 , 4 , 5 - t e t r a f l u o ro p h e n y l ) - 1 , 3 , 4 - o x a d i a z o l - 2 - y l ) - methanimine (A1): Infrared (IR) (KBr) ν cm-1: 3157, 3071 (C-H stretching, aromatic), 2988, 2921 (C-H stretching, aliphatic), 1593 (C=C stretching), 1524 (N-N stretching), 1485 (C=N stretching, oxadiazole), 1448 (C=N stretching, diazomethane), 1356 (C-H bending, SCH3), 1247 (C-N stretching), 1142 (C-O-C stretching), 1093 (C-F stretching), 1003 (C-H out of plane), 812 (C-H bending, diazomethane), 754 (C-S stretching); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.93 (s, 1H, N=CH), 7.98 (s, 1H, tetrafluorophenyl), 7.60 (d, 2H, J=5.36 Hz, methyl(phenyl)sulfide), 7.30 (d, 2H, J=8.48 Hz, methyl(phenyl)sulfide), 2.49 (s, 3H, SCH3).
N-(3-((4-(methylthio)benzylidene)amino)-5- (2,3,4,5-tetrafluorophenyl)-4H-1,2,4-triazol-4- yl)isonicotinamide (B1): IR (KBr) ν cm-1: 3393 (N-H stretching), 3159, 3067 (C-H stretching, aromatic), 2913, 2871 (C-H stretching, aliphatic), 1753 (C=O stretching), 1667 (N-H bending), 1597 (C=C stretching), 1573 (N-N stretching), 1491 (C=N stretching, oxadiazole), 1448 (C=N stretching, diazomethane), 1351 (C-H bending, SCH3), 1253 (C-N stretching), 1097 (C-F stretching), 1009 (C-H out of plane), 824 (C-H bending, diazomethane), 750 (C-S stretching); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.09 (s, 1H, N=CH), 7.78 (d, 2H, J=4.64 Hz, isoniazid), 7.56 (d, 2H, J=8.40 Hz, methyl(phenyl) sulfide), 7.37 (d, 2H, J=8.96 Hz, methyl(phenyl) sulfide), 7.17 (d, 2H, J=8.40 Hz, isoniazid), 6.66 (s, 1H, N-H), 2.45 (s, 3H, SCH3); 13C NMR (400 MHz, DMSO-d6) δ (ppm): 161.24 (N=CH), 156.79 (C1 triazole), 153.06, 152.88 (C=O), 151.92, 151.42 (C2 triazole), 146.97, 139.22, 137.80, 131.22, 129.53, 126.71, 125.47, 113.12, 108.82, 14.57 (SCH3); Electrospray Ionisation Mass Spectrometry (ESI-MS) (m/z): 501.46 M+, 503.46 [M+2]+ peak calculated and 502 M+, 504 [M+2]+ peak found.
N - ( 3 - p e n t a f l u o r o p h e n y l ) - 5 - ( ( ( E ) - 3 - phenylallylidene)amino)-4H-1,2,4-triazol-4-yl) isonico tinamide (B2): IR (KBr) ν cm-1: 3325 (N-H stretching), 3046 (C-H stretching, aromatic), 2964, 2913, 2889 (C-H stretching, aliphatic), 1716 (C=O stretching), 1684 (N-H bending), 1607 (C=C stretching), 1564 (N-N stretching), 1483 (C=N stretching, triazole), 1452 (C=N stretching, diazomethane), 1436 (C-H bending, CH2), 1259 (C-N stretching), 1084 (C-F stretching), 998 (C-H out of plane), 923 (C-H bending, CH2), 874 (C-H bending, diazomethane); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.80 (d, 2H, J=5.72 Hz, isoniazid), 8.72 (d, 1H, J=5.52 Hz, N=CH), 7.66 (d, 2H, J=7.12 Hz, phenyl ring), 7.60 (s, 1H, N-H), 7.53 (t, 2H, J=7.32 Hz, phenyl ring), 7.43 (t, 1H, J=7.36 Hz, phenyl ring), 7.29 (d, 1H, J=2.20 Hz, HC=CH-Ar), 7.02 (d, 2H, J=5.44 Hz, isoniazid), 6.77 (t, 1H, J=2.20 Hz, HC=CH-Ar); 13C NMR (400 MHz, DMSO-d6) δ (ppm): 163.48 (N=CH), 156.55, 154.91 (C1 triazole), 152.73 (C=O), 151.95 (C2 triazole), 149.98, 143.51, 138.36, 134.86 (HC=CH), 130.67, 128.46, 126.63 (aromatic carbons), 121.38 (HC=CH), 111.73; ESIMS (m/z): 499.40 M+ peak calculated and 499 M+ peak found.
N-(3-((4-propylbenzylidene)amino)-5-(2,3,4,5- t e t r a f l u o ro p h e n y l ) - 4 H - 1 , 2 , 4 - t r i a z o l - 4 - y l ) isonicotinamide (B3): IR (KBr) ν cm-1: 3417 (N-H stretching), 3068 (C-H stretching, aromatic), 2956, 2871 (C-H stretching, aliphatic), 1690 (C=O stretching), 1648 (N-H bending), 1603 (C=C stretching), 1506 (N-N stretching), 1443 (C=N stretching), 1357 (C-H bending, SCH3), 1254, 1138 (C-N stretching), 1091 (C-F stretching), 957 (C-H out of plane), 936 (C-H bending, CH2), 830 (C-H bending, diazomethane), 730 (C-S stretching); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.76 (s, 1H, tetrafluorophenyl), 8.12 (s, 1H, N=CH), 7.89 (d, 2H, J=4.82 Hz, isoniazid), 7.68 (d, 2H, J=8.52 Hz, propylphenyl), 7.33 (d, 2H, J=8.63 Hz, propylphenyl), 7.11 (d, 2H, J=8.36 Hz, isoniazid), 6.35 (s, 1H, N-H), 2.61 (t, 2H, J=2.58 Hz, CH2CH2CH3), 1.67 (m, 2H, J=2.86 Hz, CH2CH2CH3), 0.92 (t, 3H, J=3.16 Hz, CH3); 13C NMR (400 MHz, DMSO-d6) δ (ppm): 163.71 (N=CH), 155.33, 153.46 (C1 triazole), 152.06 (C=O), 150.96 (C2 triazole), 150.28, 143.86, 140.15, 138.76, 136.02, 134.83, 129.36, 128.23, 127.84, 120.31, 111.32 (aromatic carbons), 38.13 (CH2), 22.42 (CH2), 13.11 (CH3); ESI-MS (m/z): 497.45 M+ peak calculated and 497 M+ peak found
N - ( 3 - ( ( 4 - f l u o r o b e n z y l i d e n e ) a m i n o ) - 5 - ( p e r f l u o ro p h e n y l ) - 4 H - 1 , 2 , 4 - t r i a z o l - 4 - y l ) isonicotinamide (B4): IR (KBr) ν cm-1: 3419 (N-H stretching), 3151, 3057 (C-H stretching, aromatic), 2926, 2882 (C-H stretching, aliphatic), 1702 (C=O stretching), 1684 (N-H bending), 1607 (C=C stretching), 1565 (N-N stretching), 1449 (C=N stretching), 1258 (C-N stretching), 1101 (C-F stretching), 998 (C-H out of plane), 870 (C-H bending, diazomethane); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.16 (s, 1H, N=CH), 7.72 (d, 2H, J=4.76 Hz, isoniazid), 7.61 (d, 2H, J=8.44 Hz, fluorophenyl), 7.43 (d, 2H, J=8.12 Hz, fluorophenyl), 7.21 (d, 2H, J=8.16 Hz, isoniazid), 6.39 (s, 1H, N-H); 13C NMR (400 MHz, DMSO-d6) δ (ppm): 164.11 (N=CH), 156.21, 153.16 (C1 triazole), 151.86 (C=O), 150.27 (C2 triazole), 149.17, 143.42, 140.31, 138.64, 135.82, 133.83, 129.36, 128.54, 127.67, 119.54, 111.91; ESI-MS (m/z): Calculated 491.35 M+ peak calculated and 492 M+ peak found.
N - ( 3 - p e n t a f l u o r o p h e n y l ) - 5 - ( ( - 4 - propylbenzylidene)amino)-4H-1,2,4-triazol-4- yl) isonicotinamide (B5): IR (KBr) ν cm-1: 3413 (N-H stretching), 3056 (C-H stretching, aromatic), 2974, 2856 (C-H stretching, aliphatic), 1694 (C=O stretching), 1649 (N-H bending), 1605 (C=C stretching), 1506 (N-N stretching), 1443 (C=N stretching), 1357 (C-H bending, CH3), 1256, 1139 (C-N stretching), 1093 (C-F stretching), 957 (C-H out of plane), 940 (C-H bending, CH2), 838 (C-H bending, diazomethane); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.19 (s, 1H, N=CH), 7.93 (d, 2H, J=4.90 Hz, isoniazid), 7.66 (d, 2H, J=8.56 Hz, propylphenyl), 7.32 (d, 2H, J=8.63 Hz, propylphenyl), 7.12 (d, 2H, J=8.40 Hz, isoniazid), 6.36 (s, 1H, N-H), 2.61 (t, 2H, J=2.58 Hz, CH2CH2CH3), 1.65 (m, 2H, J=2.90 Hz, CH2CH2CH3), 0.93 (t, 3H, J=3.12 Hz, CH3); 13C NMR (400 MHz, DMSO-d6) δ (ppm): 162.94 (N=CH), 155.33, 153.49 (C1 triazole), 152.16 (C=O), 150.91 (C2 triazole), 150.38, 143.86, 140.15, 138.76, 135.83, 134.83, 129.36, 128.23, 127.84, 121.23, 111.53 (aromatic carbons), 36.43 (CH2), 21.37 (CH2), 13.18 (CH3); ESI-MS (m/z): 515.44 M+ peak calculated and 516 M+ peak found.
N-(3-((4-(methylthio)benzylidene)amino)-5- (pentafluorophenyl)-4H-1,2,4-triazol-4-yl) isonicotinamide (B6): IR (KBr) ν cm-1: 3417 (N-H stretching), 3157, 3068 (C-H stretching, aromatic), 2979, 2913 (C-H stretching, aliphatic), 1750 (C=O stretching), 1685 (N-H bending), 1597 (C=C stretching), 1573 (N-N stretching), 1489 (C=N stretching, oxadiazole), 1448 (C=N stretching, diazomethane), 1356 (C-H bending, SCH3), 1241 (C-N stretching), 1090 (C-F stretching), 1013 (C-H out of plane), 813 (C-H bending, diazomethane), 750 (C-S stretching); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.24 (s, 1H, N=CH), 7.88 (d, 2H, J=4.94 Hz, isoniazid), 7.54 (d, 2H, J=8.48 Hz, methyl(phenyl) sulfide), 7.39 (d, 2H, J=8.92 Hz, methyl(phenyl) sulfide), 7.19 (d, 2H, J=8.40 Hz, isoniazid), 6.64 (s, 1H, N-H), 2.46 (s, 3H, SCH3); 13C NMR (400 MHz, DMSO-d6) δ (ppm): 162.54 (N=CH), 156.46 (C1 triazole), 153.26, 152.78 (C=O), 151.21 (C2 triazole), 147.04, 139.32, 137.68, 130.29, 129.53, 126.67, 125.42, 113.12, 108.72 (aromatic carbons), 14.18 (SCH3); ESI-MS (m/z): 519.45 M+, 521.45 [M+2]+ peak calculated and 520 M+, 522 [M+2]+ peak found.
N-(3-((4-Fluorobenzylidene)amino)-5-(2,3,4,5- t e t r a f l u o ro p h e n y l ) - 4 H - 1 , 2 , 4 - t r i a z o l - 4 - y l ) isonicotinamide (B7): IR (KBr) ν cm-1: 3358 (N-H stretching), 3162, 3051 (C-H stretching, aromatic), 2937, 2867 (C-H stretching, aliphatic), 1712 (C=O stretching), 1652 (N-H bending), 1612 (C=C stretching), 1564 (N-N stretching), 1454 (C=N stretching), 1257 (C-N stretching), 1103 (C-F stretching), 996 (C-H out of plane), 874 (C-H bending, diazomethane); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.84 (s, 1H, tetraflurophenyl), 8.18 (s, 1H, N=CH), 7.76 (d, 2H, J=4.76 Hz, isoniazid), 7.58 (d, 2H, J=8.36 Hz, fluorophenyl), 7.45 (d, 2H, J=8.16 Hz, fluorophenyl), 7.19 (d, 2H, J = 8.20 Hz, isoniazid), 6.42 (s, 1H, N-H); 13C NMR (400 MHz, DMSO-d6) δ (ppm): 163.27 (N=CH), 156.23 (C1 triazole), 154.12, 152.39 (C=O), 151.46 (C2 triazole), 146.83, 139.68, 137.54, 130.37, 129.26, 126.34, 125.21, 112.86, 109.17; ESI-MS (m/z): 473.36 M+ peak calculated and 473 M+ peak found.
N - ( 3 - ( ( ( E ) - 3 - p h e n y l a l l y l i d e n e ) a m i n o ) - 5 - (2,3,4,5-tetrafluorophenyl)-4H-1,2,4-triazol-4- yl)isonicotinamide (B8): IR (KBr) ν cm-1: 3339 (N-H stretching), 3045 (C-H stretching, aromatic), 2968, 2918, 2894 (C-H stretching, aliphatic), 1720 (C=O stretching), 1680 (N-H bending), 1607 (C=C stretching), 1564 (N-N stretching), 1482 (C=N stretching, triazole), 1446 (C=N stretching, diazomethane), 1436 (C-H bending, CH2), 1260 (C-N stretching), 1084 (C-F stretching), 998 (C-H out of plane), 922 (C-H bending, CH2), 872 (C-H bending, diazomethane); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.88 (s, 1H, tetraflurophenyl), 8.76 (d, 2H, J=5.68 Hz, isoniazid), 8.71 (d, 1H, J=5.56 Hz, N=CH), 7.64 (d, 2H, J=7.16 Hz, phenyl ring), 7.54 (t, 2H, J=7.28 Hz, phenyl ring), 7.45 (t, 1H, J=7.40 Hz, phenyl ring), 7.31 (d, 1H, J=2.16 Hz, HC=CH-Ar), 7.06 (d, 2H, J=5.40 Hz, isoniazid), 6.77 (t, 1H, J=2.24 Hz, HC=CH-Ar), 6.52 (s, 1H, N-H); 13C NMR (400 MHz, DMSO-d6) δ (ppm): 163.37 (N=CH), 156.33, 153.77 (C1 triazole), 152.67 (C=O), 151.62 (C2 triazole), 150.39, 143.25, 138.36, 134.83 (HC=CH), 130.56, 128.46, 126.53, 121.34 (HC=CH), 111.87; ESI-MS (m/z): 481.40 M+ peak calculated and 481 M+ peak found.
N-(3-((4-chlorobenzylidene)amino)-5-(2,3,4,5- t e t r a f l u o ro p h e n y l ) - 4 H - 1 , 2 , 4 - t r i a z o l - 4 - y l ) isonicotinamide (B9): IR (KBr) ν cm-1: 3375 (N-H stretching), 3036 (C-H stretching, aromatic), 2964, 2873 (C-H stretching, aliphatic), 1712 (C=O stretching), 1678 (N-H bending), 1598 (C=C stretching), 1563 (N-N stretching), 1443 (C=N stretching), 1269 (C-N stretching), 1110 (C-F stretching), 993 (C-H out of plane), 838 (C-H bending, diazomethane), 815 (C-Cl stretching); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.74 (s, 1H, tetrafluorophenyl), 8.18 (s, 1H, N=CH), 7.75 (d, 2H, J=4.72 Hz, isoniazid), 7.64 (d, 2H, J=8.52 Hz, chlorophenyl), 7.47 (d, 2H, J=7.96 Hz, chlorophenyl), 7.17 (d, 2H, J=8.12 Hz, isoniazid), 6.42 (s, 1H, N-H); 13C NMR (400 MHz, DMSO-d6) δ (ppm): 161.97 (N=CH), 155.82, 153.42 (C1 triazole), 152.27 (C=O), 151.19 (C2 triazole), 150.26, 143.15, 138.24, 130.31, 128.46, 126.13, 111.32; ESI-MS (m/z): 489.81 M+ peak calculated and 490 M+ peak found.
N - ( 3 - ( ( 4 - c h l o r o b e n z y l i d e n e ) a m i n o ) - 5 - (pentafluorophenyl)-4H-1,2,4-triazol-4-yl) isonicotinamide (B10): IR (KBr) ν cm-1: 3396 (N-H stretching), 3034 (C-H stretching, aromatic), 2951, 2872 (C-H stretching, aliphatic), 1716 (C=O stretching), 1658 (N-H bending), 1593 (C=C stretching), 1556 (N-N stretching), 1446 (C=N stretching), 1273 (C-N stretching), 1105 (C-F stretching), 994 (C-H out of plane), 848 (C-H bending, diazomethane), 818 (C-Cl stretching); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.16 (s, 1H, N=CH), 7.71 (d, 2H, J=4.80 Hz, isoniazid), 7.63 (d, 2H, J=8.56 Hz, chlorophenyl), 7.49 (d, 2H, J=7.96 Hz, chlorophenyl), 7.15 (d, 2H, J=8.08 Hz, isoniazid), 6.41 (s, 1H, N-H); 13C NMR (400 MHz, DMSO-d6) δ (ppm): 162.67 (N=CH), 155.21, 153.26 (C1 triazole), 152.17 (C=O), 151.42 (C2 triazole), 150.67, 143.64, 138.46, 130.23, 128.67, 126.34, 111.33; ESI-MS (m/z): 507.80 M+ peak calculated and 508 M+ peak found.
Results and Discussion
Compounds were synthesized in two steps which first intermediate (E)-N-(5-(2,3,4,5-tetrafluoro- 6-substitutedphenyl)-1,3,4-oxadiazol-2-yl) substitutedimine (A1-A10) were synthesized from 2,3,4,5-tetrafluoro-6-substitutedbenzoic acid using Lewis acid as catalyst, which held on Perkin-Elmer apparatus and obtained intermediates were spectrally evaluated from IR and 1H NMR spectra. IR is given band at 1485 cm-1 (Oxdiazole C=N), 1448 cm-1 (diazomethane C=N) and 1093 cm-1 (C-O-C) and 1H NMR is given a singlet signal at 8.93 δ ppm (N=CH) which are sufficient for identification of intermediates. Final compounds 1-(3-(substitutidene)amino)-5- (2,3,4,5-tetrafluoro-6-substitutedphenyl)-4H-1,2,4- triazol-4-yl)-3-(pyridin-4-yl) urea were synthesized from intermediates using isoniazid and KOH as a catalyst. Obtained compounds characterized from IR, 1H NMR, 13C NMR and Mass spectrum. In IR spectrum, bend obtained at 1271-1245 cm-1 for (CN) instead of 1093 cm-1 (C-O-C) to indicate the formation of the final compound. In 1H NMR, signals obtained singlet at 10.10-10.30 δ ppm and 6.20- 6.45 δ ppm for (N-H). 13C NMR gives the signal at 161.10-161.40 δ ppm for (N=CH), 156.79 δ ppm for (C1 triazole) and 151.62 δ ppm for (C2 triazole). Both step products were purified by methanol and solvents used in reaction were distilled out and dried it using dry sieves as the usual manner.
Compounds were evicted for antimicrobial susceptibility. The broth dilution method was used to quantitative measure of the in vitro antimicrobial activity[23-25]. Compounds were tested against two gram positive bacterial strains; Staphylococcus aureus (MTCC-96) and Streptococcus pyogenes (S. pyogenus) (MTCC-443), two gram-negative bacterial strains; Escherichia coli (E. coli) (MTCC-442) and Pseudomonas aeruginosa (P. aeruginosa) (MTCC- 441), and for fungi, three species, Candida albicans (C. albicans) (MTCC-227), Aspergillus niger (MTCC- 282), and Aspergillus clavatus (MTCC-1323) are used. Institute of Microbial Technology, Chandigarh is procured the strains and species. To evaluating the activity of compounds against microorganisms, Minimum Inhibitory Concentrations (MICs) were measured which is the lowest concentration of the assayed antimicrobial agent to inhibits the visible growth of the microorganism tested and its expressed in μg/ml and DMSO is used as diluents/vehicles. Chloramphenicol, ciprofloxacin and norfloxacin are used as standard drug. The results depicted in Table 3 revealed that compounds B8 active against E. coli, B9 against P. aeruginosa, B2 and B7 against S. pyogenus. From the above active compounds B2 contains extended conjugation and B7, B8, B9 having electron withdrawing groups which indicate electron withdrawing group containing substituents and extending conjugation very effective against the antibacterial species. The antifungal screening data displayed in Table 3 which gives the variable inhibitory effects against fungal species. Greseofulvin and nystatin are used as a standard antifungal drug. From the screened compounds B4, B10 activity against C. albicans, which are having electron withdrawing groups. Drug susceptibility and MICs determination of the compounds against M. tuberculosis H37Rv were performed by the by L-J medium agar micro dilution method[26-28]. Rifampicin and isoniazid are used as standard drug. The MIC levels of screened compounds (B1-B10) against M. tuberculosis are given in Table 4 which explained that the compounds, showen variable inhibitory effects on the growth of the tested M. tuberculosis H37Rv strains. From the tested compounds, B2 having extended conjugation and B4 having electron withdrawing groups, which both are increase the electrone density of compounds so that they shown active against M. tuberculosis H37Rv.
| Code | Minimal Bactericidal Concentration (µg/ml) | Minimal Fungicidal Concentration (µg/ml) | |||||
|---|---|---|---|---|---|---|---|
| E. coli | P. aeruginosa | S. aureus | S. pyogenus | C. albicans | A. niger | A. clavatus | |
| MTCC 443 | MTCC 1688 | MTCC 96 | MTCC 442 | MTCC 227 | MTCC 228 | MTCC 1323 | |
| B1 | 250 | 62.5 | 125 | 250 | >1000 | >1000 | 1000 |
| B2 | 250 | 100 | 150 | 25 | 500 | 500 | 500 |
| B3 | 500 | 150 | 100 | 500 | 500 | 500 | 400 |
| B4 | 500 | 500 | 500 | 200 | 200 | 200 | 250 |
| B5 | 125 | 250 | 150 | 100 | 500 | 500 | 250 |
| B6 | 250 | 100 | 125 | 250 | 500 | 500 | 250 |
| B7 | 125 | 100 | 150 | 12.5 | 1000 | 1000 | 1000 |
| B8 | 50 | 125 | 250 | 125 | >1000 | >1000 | 500 |
| B9 | 250 | 12.5 | 100 | 62.5 | 1000 | 1000 | 500 |
| B10 | 100 | 62.5 | 250 | 200 | 250 | 250 | 250 |
| Drug | Micromolar (µg/ml) | ||||||
| Chloramphenicol | 50 | 50 | 50 | 50 | - | - | - |
| Ciprofloxacin | 25 | 25 | 50 | 50 | - | - | - |
| Norfloxacin | 10 | 10 | 10 | 10 | - | - | - |
| Nystatin | - | - | - | - | 100 | 100 | 100 |
| Greseofulvin | - | - | - | - | 500 | 100 | 100 |
Note: All the values are presented as mean of six experiments. Antimicrobial activity is zero for 2P DMSO which used as control and diluent
Table 3: Antimicrobial Activity Data of Compounds (B1-B10)
| H37Rv MIC in µg/ml | Discriptors Used | ||||
|---|---|---|---|---|---|
| Code | Actual | Actual | Predicted | Residual | XA Most Hydrophilic |
| MIC | pMIC | pMIC | pMIC | ||
| B1 | 125 | -2.09 | -2.04 | 0.05 | -7.5752 |
| B2 | 25 | -1.39 | -1.45 | -0.06 | -7.5750 |
| B3 | 100 | -2.00 | -2.04 | -0.04 | -7.5752 |
| B4 | 12.5 | -1.09 | -1.15 | -0.06 | -7.5749 |
| B5 | 100 | -2.00 | -1.74 | -0.26 | -7.5751 |
| B6 | 100 | -2.00 | -1.74 | -0.26 | -7.5754 |
| B7 | 50 | -1.69 | -1.45 | -0.24 | -7.5750 |
| B8 | 62.5 | -1.79 | -1.74 | 0.05 | -7.5749 |
| B9 | 125 | -2.09 | -2.33 | -0.14 | -7.5753 |
| B10 | 62.5 | -1.79 | -1.74 | -0.05 | -7.5749 |
| Drug | Micromolar (µg/ml) | ||||
| Isoniazid | 0.20 | ||||
| Rifampicin | 40 | ||||
Note: pMIC=(1/log MIC) value used for 2D-QSAR determination. All the values of MIC presented as mean of six experiments. Antitubercular activity is zero for 2P DMSO which used as control and diluent
Table 4: Antitubercular Activity and 2D-QSAR Data of The Compounds (B1-B10)
SAR study suggested that 3, 4, and 5 substituted 1,2,4-triazole is required for more potent antimicrobial and antitubercular activity[29,30]. At Position-3 HC=N Schiff base and aromatic ring are more favorable to increase the antimicrobial and antitubercular activity. At position-4 isoniazid increased the antitubercular activity and at position-5 aromatic ring increased the antimicrobial activity due to increased resonance. 1,2,4-Triazole is itself increased the activity due to double bonds between carbon and nitrogen atom. The arrangement of the substitution position and groups are also effective to increase the biological effects. Resonance with 1,2,4-triazole derivatives are given optimum activity against antimicrobial species. Position-3 contains C=N Schiff base and electron withdrawing group having aromatic substitution which favorable to increase the biological activity. Toxophoric C=N linkage of Schiff base is increased the antimicrobial activity[31]. Isoniazid is itself an antitubercular drug and its derivatives are potent antitubercular activity against M. tuberculosis H37Rv (MTB) and multi-drug resistant M. tuberculosis (MDR-TB)[32]. Fluorinated compounds are given the potent antimicrobial activity. A fluorine atoms are increased the pharmacological properties due to more electronegativity, low polarizability, small atom size and strong carbon-fluorine bond. Cinnamal phenyl have exhausted more effective activity due to extending conjugation. The arrangement of groups and rings also affects better to good biological activities[33]. We are further process the in vivo activity for more potent compounds.
2D-QSAR analysis is used to understand observed biological properties and determines the structural parameter control biological activity. A large number of molecular descriptors (physicochemical, spatial, electronic and topological parameters) are usually used in the QSAR analysis using Vlife MDS software. The various physicochemical descriptors calculated the free energy change due to drug receptor complex and topological structure descriptors are used for alignment independent descriptor, which both independent descriptors are considered as independent variables. Spatial parameters are given the steric features of the drug molecules which required to fit with the receptor. Non-covalent bonding between the drug molecules and the receptors, which is described the electronic descriptors. QSAR analysis regression was performed using pMIC values as dependent variables and calculated parameters as independent variables. Manually selecting and placing molecules in the training and test sets comprising of 8 and 2 molecules, respectively.
PLSR method is generated significant QSAR model[34], which is considered on the basis of statistical parameters, correlation coefficient (r), squared correlation coefficient (r2), predictive r2 for external test set (pred r2) for external validation, and Fischer’s value (F). External validation for the activity of the test set was predicted using the model developed by the training set[35]. The cross-validated coefficient, q2 was calculated[36]. The significance of the models, hence obtained is derived based on a calculated Z score[37].
pMIC (pMIC=log(1/MIC)) values are applied for the 2D-QSAR prediction of M. tuberculosis H37Rv. PLSR methodology was used up for the 2D-QSAR prediction of the compounds from VLife MDS software, which counting the term selection criterion as r2, q2, pred_r2 and F test. The training and test sets of compounds were selected by the sphere exclusion method and the models were validated by both internal and external validation procedures. The model gives the following equation for pMIC prediction.
pMIC=+2957.0432
XAMostHydrophilic+22398.1532
Ntraining=5, Ntest=5, Degree of freedom=6, r2=0.8940, q2=0.8197, F test=50.6234, r2_se=109.5612, q2_ se=142.9109, pred_r2=0.6675, pred_r2se=367.2893
The equation explains 89 % (r2=0.8940) of the total variance in the training set as well as it has internal (q2) and external (pred_r2) predictive ability of ~81 % and ~66 % respectively. The F test=50.6234 shows the statistical significance of the model. The model is incorporated with XA Most Hydrophilic parameters with their corresponding values for each molecule in the selected model given in Table 3. The descriptor XA Most Hydrophilic in the model is indices the highest hydrophilicity of the compounds correlate with the antitubercular activity. The positive correlation suggests that antitubercular activity directly proportional to highest hydrophilicity of 1,2,4-triazole derivatives. The model is validated by ZScore R^2=5.53757, ZScore Q^2=3.57107, Best Rand R^2=0.40359, Best Rand Q^2=-0.00449, Alpha Rand R^2=0.0000, Alpha Rand Q^2=0.0050, ZScore Pred R^2=1.57248, best Rand Pred R^2=0.33061, Alpha Rand Pred R^2=0.1000. The randomization test suggests that the developed model has a probability of less than 1 % that the model is generated by chance.
The observed MIC values are near to predicted MIC values. Predicted values, Residual values and used descriptors shown in Table 3. Fitness graph Predicted vs Actual data given in graph.
It is mandatory to study the pharmacokinetics properties, i.e., absorption in the body, distribution into the different compartments, metabolism by organs and elimination through the body. Computational studies of the ADME parameters are mandatory to designed the molecules which prioritize for synthesis[38]. Hence, the ADME study is an essential step for checking the drug-likeness. ADME studies of the synthesised compounds (C1-C10) were carried out using the Swiss ADME tool[39]. QSAR and druglikeness are also predicted an octanol/water partition coefficient (log Po/w), Topological Polar Surface Area (TPSA), Hydrogen Bond Acceptor (HBA), Hydrogen Bond Donor (HBD), Lipinski Rule and synthetic accessibility which are tabulated in Table 5.
| Code | RB | HBA | HBD | MR | Log S | GI absorption and BBB Permeant | Log Kp (cm/s) | SA | TPSA | Lipinski Rule |
|---|---|---|---|---|---|---|---|---|---|---|
| B1 | 6 | 9 | 1 | 111.70 | -5.10 | High and no | -6.45 | 3.72 | 85.06 | 1 |
| B2 | 7 | 10 | 1 | 116.62 | -5.29 | Low and no | -6.52 | 3.81 | 85.06 | 1 |
| B3 | 8 | 9 | 1 | 121.31 | -9.46 | Low and no | -5.93 | 3.88 | 85.06 | 1 |
| B4 | 6 | 7 | 1 | 106.65 | -5.12 | Low and no | -6.71 | 3.61 | 85.06 | 1 |
| B5 | 8 | 10 | 1 | 121.27 | -5.89 | Low and no | -5.97 | 3.88 | 85.06 | 2 |
| B6 | 6 | 9 | 1 | 111.70 | -5.10 | High and no | -6.45 | 3.72 | 85.06 | 1 |
| B7 | 6 | 10 | 1 | 106.69 | -4.96 | High and no | -6.67 | 3.61 | 85.06 | 1 |
| B8 | 7 | 9 | 1 | 116.67 | -5.13 | High and no | -6.48 | 3.81 | 85.06 | 1 |
| B9 | 6 | 9 | 1 | 111.74 | -5.39 | High and no | -6.39 | 3.6 | 85.06 | 1 |
| B10 | 6 | 10 | 1 | 111.70 | -5.55 | Low and no | -6.43 | 3.6 | 85.06 | 1 |
Note: RB: Rotatable Bond, HBA: Hydrogen Bond Acceptors, HBD: Hydrogen Bond Donors, MR: Molar Refractive, SA: Synthetic Accessibility, TPSA: Topologocal Polar Surface Area
Table 5: In Silico Admet Properties Data of The Compounds (B1-B10)
Lipinski rule of five is given by Lipinski et al.[40] in 1997, the rule of five is based on exact norms to predict the drug-likeness of a molecule which having a pharmacological activities. These norms are log P<5, number of HBD<5, HBA<10 and M.W≤500 Da. This rule is used in preselect molecules which presenting good ADME properties that must have a medicament in the organism. Here, we are used the SwissADME property calculator (http://www.SwissADME.com) to measured the four parameters of Lipinski’s rule. The number of rotatable bonds that are to be lesser than 10 have a good oral bioavailability[41].
In conclusion, biologically active isoniazid and Schiff bases having 1,2,4-triazole derivatives 1-(3-(Substitutidene)amino)-5-(2,3,4,5-tetrafluoro- 6-substitutedphenyl)-4H-1,2,4-triazol-4-yl)-3- (pyridin-4-yl)urea was synthesized. These derivatives are contained antimicrobial and antitubercular active toxophores isoniazid, 1,2,4-triazole and Schiff base. From this toxophoric unit isoniazid is increased antitubercular activity while 1,2,4-triazole and Schiff base increased the antimicrobial activity. The derivatives also having the fluorinated molecules which are increased the biological activity of compounds because of its electron withdrawing nature. From the biological results, we concluded an electron withdrawing group containing substituents and extending conjugation increased the biological activity. We are also performed the 2D-QSAR for M. tuberculosis H37Rv using VLife MDS software which suggested that antitubercular activity of the compounds are directly proportional to most hydrophilicity of the compounds.
Acknowledgments:
Authors are thankful to Department of Chemistry, Veer Naemad South Gujarat University, Surat, India for providing laboratory facilities and thankful to SAIF Chandigarh for spectral analysis as well as thankful to Microcare laboratory for Biological study. One of the author Jaydeep A. Patel is thankful to the University Grant Commission, India for National Fellowship for Higher Education to providing funding sources.
Funding:
National Fellowship for Higher Education (NFHE) of ST Student, University Grant Commission, India. Award Number: F1-17.1/2016-17/NFST-2015-17- ST-GUJ-2209.
Conflict of interest:
The authors have no conflict of interest to declare.
References
- Liu P, Zhu S, Li P, Xie W, Jin Y, Sun Q et al. Synthesis and SAR studies of biaryloxy-substituted triazoles as antifungal agents. Bioorgan Med Chem Lett 2008;18(11):3261-5.
[Crossref] [Google Scholar] [PubMed]
- Isloor AM, Kalluraya B, Shetty P. Regioselective reaction: Synthesis, characterization and pharmacological studies of some new Mannich bases derived from 1, 2, 4-triazoles. Eur J Med Chem 2009;44(9):3784-7.
[Crossref] [Google Scholar] [PubMed]
- Shiradkar M, Kumar GV, Dasari V, Tatikonda S, Akula KC, Shah R. Clubbed triazoles: A novel approach to antitubercular drugs. Eur J Med Chem 2007;42(6):807-16.
[Crossref] [Google Scholar] [PubMed]
- Mishra N, Arora P, Kumar B, Mishra LC, Bhattacharya A, Awasthi SK, Bhasin VK. Synthesis of novel substituted 1, 3-diaryl propenone derivatives and their antimalarial activity in vitro. Eur J Med Chem 2008;43(7):1530-5.
[Crossref] [Google Scholar] [PubMed]
- Ahmad A, Varshney H, Rauf A, Sherwani A, Owais M. Synthesis and anticancer activity of long chain substituted 1, 3, 4-oxadiazol-2-thione, 1, 2, 4-triazol-3-thione and 1, 2, 4-triazolo [3, 4-b]-1, 3, 4-thiadiazine derivatives. Arabian J Chem 2017;10(2):S3347-57.
- Kuş C, Ayhan-Kılcıgil G, Özbey S, Kaynak FB, Kaya M, Çoban T, et al. Synthesis and antioxidant properties of novel N-methyl-1, 3, 4-thiadiazol-2-amine and 4-methyl-2H-1, 2, 4-triazole-3 (4H)-thione derivatives of benzimidazole class. Bioorgan Med Chem 2008;16(8):4294-303.
[Crossref] [Google Scholar] [PubMed]
- El-Gazzar YI, Georgey HH, El-Messery SM, Ewida HA, Hassan GS, Raafat MM, et al. Synthesis, biological evaluation and molecular modeling study of new (1, 2, 4-triazole or 1, 3, 4-thiadiazole)-methylthio-derivatives of quinazolin-4 (3H)-one as DHFR inhibitors. Bioorgan Chem 2017;72:282-92.
- Abosadiya HM, Abusaadiya SM, Hasbullah SA, Yamin BM. Synthesis, characterization, crystal structures and DFT studies of some new 1, 2, 4-triazole and triazolidin derivatives. J Mol Struct 2018;1151:315-26.
- Tseng WC, Wang LY, Wu TS, Wong FF. ‘One-flask’synthesis to 3, 5-disubstituted 1, 2, 4-triazoles from aldehydes with hydrazonoyl hydrochlorides via 1, 3-dipolar cycloaddition. Tetrahedron 2011;67(29):5339-45.
- Banerjee AG, Das N, Shengule SA, Sharma PA, Srivastava RS, Shrivastava SK. Design, synthesis, evaluation and molecular modelling studies of some novel 5, 6-diphenyl-1, 2, 4-triazin-3 (2H)-ones bearing five-member heterocyclic moieties as potential COX-2 inhibitors: A hybrid pharmacophore approach. Bioorgan Chem 2016;69:102-20.
[Crossref] [Google Scholar] [PubMed]
- Poonia K, Siddiqui S, Arshad M, Kumar D. In vitro anticancer activities of Schiff base and its lanthanum complex. Spectrochim Acta Part A 2016;155(15):146-54.
- Jia L, Xu J, Zhao X, Shen S, Zhou T, Xu Z, et al. Synthesis, characterization, and antitumor activity of three ternary dinuclear copper (II) complexes with a reduced Schiff base ligand and diimine coligands in vitro and in vivo. J Inorgan Biochem 2016;159:107-19.
[Crossref] [Google Scholar] [PubMed]
- Ferreira MD, Vasconcelos TR, de Carvalho EM, Lourenço MC, Wardell SM, Wardell JL, et al. Synthesis and antitubercular activity of novel Schiff bases derived from D-mannitol. Carbohydrate Res 2009;344(15):2042-7.
- Mishra N, Poonia K, Soni SK, Kumar D. Synthesis, characterization and antimicrobial activity of Schiff base Ce (III) complexes. Polyhedron 2016;120(14):60-8.
- Kulkarni AA, Wankhede SB, Dhawale ND, Yadav PB, Deore VV, Gonjari ID. Synthesis, characterization and biological behavior of some Schiff's and Mannich base derivatives of Lamotrigine. Arabian J Chem 2017;10:S184-9.
- Harries AD, Dye C. Tuberculosis. Ann Trop Med Parasitol 2006;100:415-31.
- World Health Organization, World Health Organization (WHO) Report, 2003.
- Nawale VV, Bhandari S, Raut KG, Wankhade PP. Design of potential antitubercular agents containing urea analogues, using pharmacophore optimization by molecular modeling studies. Indo Am J Pharm Sci 2018;5(1):395-412.
- Aggarwal N, Kumar R, Dureja P, Khurana JM. Synthesis, antimicrobial evaluation and QSAR analysis of novel nalidixic acid based 1, 2, 4-triazole derivatives. Eur J Med Chem 2011;46(9):4089-99.
[Crossref] [Google Scholar] [PubMed]
- Martins F, Santos S, Ventura C, Elvas-Leitão R, Santos L, Vitorino S, et al. Design, synthesis and biological evaluation of novel isoniazid derivatives with potent antitubercular activity. Eur J Med Chem 2014;81:119-38.
[Crossref] [Google Scholar] [PubMed]
- Nitin M, Jyoti A, Dheeraj A, Pankaj M, Tanaji M, Sivakumar T. Synthesis, antimicrobial and anti-inflammatory activity of some 5-substituted-3-pyridine-1, 2, 4-triazoles. Int J Pharm Tech Res 2010;2(4):2450-5.
- Singh R, Kashaw SK, Mishra VK, Mishra M, Rajoriya V, Kashaw V. Design and synthesis of new bioactive 1, 2, 4-triazoles, potential Antitubercular and antimicrobial agents. Indian J Pharm Sci 2018;80(1):36-45.
- Al-Bakri AG, Afifi FU. Evaluation of antimicrobial activity of selected plant extracts by rapid XTT colorimetry and bacterial enumeration. J Microbiol Methods 2007;68(1):19-25.
[Crossref] [Google Scholar] [PubMed]
- Liang H, Xing Y, Chen J, Zhang D, Guo S, Wang C. Antimicrobial activities of endophytic fungi isolated from Ophiopogon japonicus (Liliaceae). BMC Complem Altern Med 2012;12:238-43.
[Crossref] [Google Scholar] [PubMed]
- Monteiro MC, de la Cruz M, Cantizani J, Moreno C, Tormo JR, Mellado E, et al. A new approach to drug discovery: High-throughput screening of microbial natural extracts against Aspergillus fumigatus using resazurin. J Biomol Screen 2012;17(4):542-9.
[Crossref] [Google Scholar] [PubMed]
- Canetti G, Froman S, Grosset JA, Hauduroy P, Langerova M, Mahler HT, et al. Mycobacteria: Laboratory methods for testing drug sensitivity and resistance. Bull World Health Organ 1963;29(5):565.
- Sánchez JG, Kouznetsov VV. Antimycobacterial susceptibility testing methods for natural products research. Brazil J Microbiol 2010;41(2):270-7.
- Seethala R, Zhang L. Handbook of drug screening. 2nd ed. New York: CRC Press;2009.
- Seelam N, Shrivastava SP, Prasanthi S, Gupta S. Synthesis and in vitro study of some fused 1, 2, 4-triazole derivatives as antimycobacterial agents. J Saudi Chem Soc 2016;20(4):411-8.
- Mange YJ, Isloor AM, Malladi S, Isloor S, Fun HK. Synthesis and antimicrobial activities of some novel 1, 2, 4-triazole derivatives. Arab J Chem 2013;6(2):177-81.
- Matar SA, Talib WH, Mustafa MS, Mubarak MS, AlDamen MA. Synthesis, characterization, and antimicrobial activity of Schiff bases derived from benzaldehydes and 3, 3′-diaminodipropylamine. Arab J Chem 2015;8(6):850-7.
[Crossref] [Google Scholar] [PubMed]
- Manjashetty TH, Yogeeswari P, Sriram D. Microwave assisted one-pot synthesis of highly potent novel isoniazid analogues. Bioorg Med Chem Lett 2011;21(7):2125-8.
[Crossref] [Google Scholar] [PubMed]
- Tuncbilek M, Kiper T, Altanlar N. Synthesis and in vitro antimicrobial activity of some novel substituted benzimidazole derivatives having potent activity against MRSA. Eur J Med Chem 2009;44(3):1024-33.
[Crossref] [Google Scholar] [PubMed]
- Sharma MC, Sharma S, Sahu NK, Kohli DV. QSAR studies of some substituted imidazolinones angiotensin II receptor antagonists using Partial Least Squares Regression (PLSR) method based feature selection. J Saudi Chem Soc 2013;17(2):219-25.
[Crossref] [Google Scholar] [PubMed]
- Desai NC, Kotadiya GM, Trivedi AR, Khedkar VM, Jha PC. Design, synthesis, and biological evaluation of novel fluorinated pyrazole encompassing pyridyl 1, 3, 4-oxadiazole motifs. Med Chem Res 2016;25:2698-717.
- Noolvi MN, Patel HM. A comparative QSAR analysis and molecular docking studies of quinazoline derivatives as tyrosine kinase (EGFR) inhibitors: A rational approach to anticancer drug design. J Saudi Chem Soc 2013;17(4):361-79.
- Sharma MC. 2D QSAR studies of the inhibitory activity of a series of substituted purine derivatives against c-Src tyrosine kinase. J Taibah Univ Sci 2016;10(4):563-70.
- Nagaladinne N, Hindustan AA, Nayakanti D. Synthesis, characterization and docking studies of N-methyl-2, 3-Dihydro quinazolin-4-ones linked 1, 3-thiazole hybrids as potent anti-tubercular agents. Indian J Pharm Sci 2020;82(6):984-95.
- Daina A, Michielin O, Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017;7(1):42717.
[Crossref] [Google Scholar] [PubMed]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 1997;23(1-3):3-25.
[Crossref] [Google Scholar] [PubMed]
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002;45(12):2615-23.
[Crossref] [Google Scholar] [PubMed]