- Corresponding Author:
- T. Pongjanyakul
Department of Pharmaceutical Technology, Faculty of Pharmaceutical Sciences, Khon Kaen University, Khon Kaen-40002,Thailand
E-mail: thaned@kku.ac.th
| Date of Submission | 20 December 2011 |
| Date of Revision | 06 August 2012 |
| Date of Acceptance | 09 August 2012 |
| Indian J Pharm Sci, 2012, 74 (4): 292-301 |
Abstract
The objective of the present study was to investigate the use of propranolol-magnesium aluminium silicate intercalated complexes as drug reservoirs in hydroxypropylmethylcellulose tablets. The matrix tablets containing the complexes were prepared and characterised with respect to propranolol release and were subsequently compared with those loading propranolol or a propranolol-magnesium aluminium silicate physical mixture. Additionally, the effects of varying viscosity grades of hydroxypropyl methylcellulose, compression pressures and calcium acetate incorporation on the drug release characteristics of the complex-loaded tablets were also examined. The results showed that the complex-loaded tablets have higher tablet hardness than those containing propranolol or a physical mixture. The drug release from the complex-loaded tablets followed a zero-order release kinetic, whereas an anomalous transport was found in the propranolol or physical mixture tablets. The drug release rate of the complex tablet significantly decreased with increasing hydroxypropylmethylcellulose viscosity grade. Increase in the compression pressure caused a decrease in the drug release rate of the tablets. Furthermore, the incorporation of calcium ions could accelerate propranolol release, particularly in acidic medium, because calcium ions could be exchanged with propranolol molecules intercalated in the silicate layers of magnesium aluminium silicate. These findings suggest that propranolol-magnesium aluminium silicate intercalated complexes show strong potential for use as drug reservoirs in matrix tablets intended for modifying drug release.
Keywords
Complexes, drug release, hydroxypropylmethylcellulose, propranolol, magnesium aluminium silicate, matrix tablets
Complexation between drugs and biocompatible materials has been used for the improvement of drug solubility, stability, absorption and bioavailability [1]. In recent years, clays and biocompatible inorganic materials have been applied to adsorb the drugs onto their structures because they have large specific surface areas, good adsorption abilities and cation exchange capacities [2,3]. Magnesium aluminium silicate (MAS), a mixture of natural montmorillonite and saponite clays [4], has a layered structure that is composed of tetrahedrally coordinated silica atoms that are fused into an edge?shared octahedral plane of either aluminium hydroxide or magnesium hydroxide [4,5]. The negatively charged faces on the silicate layers of MAS form intercalated complexes through strong electrostatic interactions with amine drugs; the drugs intercalate into the silicate layers of MAS [6?8], and thereby lead to a prolonged release of the drug. This finding recently led to the potential application of drug–MAS complexes as drug carriers in matrix tablets for buccal delivery [9].
Propranolol hydrochloride (PPN), a secondary amine compound, was the first b?adrenoceptor blocking drug to achieve wide therapeutic use in angina and hypertension [10]. PPN has been selected as a drug candidate for the development of sustained?release dosage forms [11?13] due to its short half life (3.9 h) [10]. However, many researchers involved in the development of the PPN sustained?release dosage forms met with problems, such as difficulty in the control of drug release due to the high aqueous solubility of PPN. These challenges led to the use of a large amount of polymer in the matrix tablets to support the sustained release of a drug with high water solubility[14]. Recently, it was shown that PPN could electrostatically interact with MAS to form intercalated complexes. The physicochemical properties of the complex particles were characterised, and the PPN–MAS complexes showed sustained release of PPN after initial burst release [8]. Thus, it is interesting that the use of the PPN–MAS complexes as drug reservoirs in hydrophilic matrix tablets may modify drug release behaviour when compared with the tablets that contain pure PPN.
Therefore, the aim of this work was to investigate the physical properties and PPN release behaviour of the matrix tablets containing PPN–MAS complexes as drug reservoirs in comparison with those containing pure PPN or a PPN–MAS physical mixture. Hydroxypropylmethylcellulose (HPMC) has been widely used as a hydrophilic matrix forming agent [14?17] and was employed in this study. Additionally, the effects of HPMC viscosity grades, compression pressures and calcium acetate incorporation on the PPN release characteristics of the PPN–MAS complex tablets were also examined.

Figure 1:Particle and surface morphology
Particle and surface morphology of magnesium aluminium silicate (MAS) (a, b) and propranolol hydrochloride (PPN)–MAS complexes (c, d) used in this study
| Tablet | Thickness(mm) | Hardness(N) | n | K (%/min0.5)H | K0×10 (%/min) | |||
|---|---|---|---|---|---|---|---|---|
| 0.1 M HCl | PB | 0.1 M HCl | PB | 0.1 M HCl | PB | |||
| Pure drug | 6.44±0.02 | 86.6±10.8 | 0.75±0.02 | 0.72±0.04 | 4.91±0.37 | 4.49±0.30 | 3.90±0.30 | 3.60±0.20 |
| (R2=0.997) | (R2=0.987) | (R2=0.981) | (R2=0.983) | (R2=0.995) | (R2=0.991) | |||
| Physical mixture | 5.83±0.02 | 131.4±4.3 | 0.64±0.02 | 0.62±0.03 | 3.76±0.30 | 2.98±0.05 | 2.20±0.20 | 1.33±0.01 |
| (R2=0.992) | (R2=0.992) | (R2=0.994) | (R2=0.998) | (R2=0.965) | (R2=0.951) | |||
| Complexes | 5.75±0.03 | 179.1±4.4 | 0.98±0.02 | 0.89±0.03 | 3.91±0.60 | 4.28±0.36 | 2.70±0.44 | 2.90±0.25 |
| (R2=0.993) | (R2=0.990) | (R2=0.959) | (R2=0.976) | (R2=0.999) | (R2=0.987) | |||
Table 1: Characteristics of hpmc tablets.
Materials and Methods
MAS (Veegum®HV) and PPN were purchased from the R.T. Vanderbilt Company, Inc., Norwalk, USA and Changzhou Yabang Pharmaceutical Co., Ltd., Jiangsu, China, respectively. HPMC in the viscosity grades of 10?20 cP (low viscosity, LV?HPMC) and 40?60 cP (medium viscosity, MV?HPMC) was purchased from Onimax Co., Ltd., Bangkok, Thailand. High viscosity grade HPMC (HV?HPMC), 80?120 cP, was obtained from S.M. Chemical Supplies Co., Ltd., Bangkok, Thailand. All other reagents used were of analytical grade and were used as received.
Preparation of PPN–MAS complexes
A 4% w/v MAS suspension was prepared using hot water, and the suspension was cooled to room temperature prior to use. Next, 25 ml of the 4% w/v MAS suspension was mixed with 25 ml of 1% w/v PPN deionised water solution in an Erlenmeyer flask. The pH of the PPN–MAS dispersion was adjusted by adding a small amount of 1 M HCl or 1 M NaOH into the flask while swirling and using a pH meter (Ion Analyzer 250, Coring, USA) to determine when the final pH of the dispersions reached 7. Then, the dispersions were incubated at 37° with shaking for 24 h to allow PPN adsorption on the MAS to equilibrate. The PPN–MAS complexes were separated from the filtrates by filtration. Then, the collected complexes were dispersed into 25 ml of the 1% w/v PPN solution in an Erlenmeyer flask, and the mixture was incubated at 37° with shaking for 24 h for the second drug loading. The double drug?loaded PPN–MAS complexes were separated, washed and dried at 50° for 24 h. The dry PPN–MAS complexes were ground using a mortar and pestle, sieved to enable the collecting of the complex particles in the size range of 125?180 μm, and stored in a desiccator before use.
Scanning electron microscopy
Particle shape and surface morphology of MAS powder and the PPN–MAS complexes were observed using scanning electron microscopy (SEM). Samples were mounted onto stubs; sputter?coated with gold in a vacuum evaporator and photographed using a scanning electron microscope (Jeol Model JSM?6400, Tokyo, Japan).
Preparation of matrix tablets
All of the tablets were prepared using the direct compression method. The PPN–MAS complex tablet was composed of 200 mg PPN–MAS complexes (equivalent to PPN 40 mg) and 600 mg HPMC, each. The complexes and HPMC were mixed in a rotomixer for 10 min; magnesium stearate (1% w/w) was then blended with the mixture for 3 min before tabletting. The mixtures were filled into 12 mm flat?faced punches and dies and then formed by applying 6.6, 8.8 or 11.0 MPa with a hydrostatic press (Model 3126, Shimadzu, Kyoto, Japan) without holding time. The tablets obtained were stored in a desiccator prior to use.
PPN tablets and PPN–MAS physical mixture tablets were prepared for drug?release comparison with the PPN–MAS complex tablets. Each PPN tablet contained 40 mg PPN and 760 mg LV?HPMC. The PPN–MAS physical mixtures were prepared by mixing PPN and MAS in the ratio of 1:4 by weight. The PPN–MAS physical mixture tablets were composed of a 200 mg physical mixture (equivalent to PPN 40 mg) and 600 mg LV?HPMC. The preparation method of these tablets followed the protocol that was mentioned above.
The effect of calcium acetate incorporation on PPN released from the PPN–MAS complex tablets was investigated in this study. The amount of calcium acetate added was 50, 100 or 150 mg in the PPN–MAS complex tablets, in which the LV?HPMC amount was reduced to 550, 500 or 450 mg, respectively. The tableting method was the same as that mentioned previously.
Evaluation of matrix tablets
The thickness of the tablets was measured using a vernier caliper (Mitutoyo, Japan). The hardness of the tablets was measured using a tablet hardness tester (VanKel VK 200, USA). Each test was performed in triplicate.
To characterise the in vitro release characteristics, the PPN release parameters of the prepared tablets were tested using a USP dissolution apparatus 1 (basket method, VanKel 7000, USA). The tablets were placed into the basket with a rotation speed of 100 revolutions/min. The release medium was 0.1 M HCl and pH 6.8 phosphate buffer in a volume of 750 ml, and the temperature was controlled at 37.0±0.5°. Twenty millilitres of each of the samples was collected and replaced with fresh medium at various time intervals. The amount of PPN released was analysed using a UV/Vis spectrophotometer (Shimadzu UV1201, Japan) at a wavelength of 290 nm. In the case of the calcium ion effect study, the release medium was a 0.1 M HCl and pH 6.8 Tris buffer containing 8.19 g/l sodium chloride and 0.32 g/l potassium chloride for simulating the sodium and potassium ions present in the small intestine [18]. Tris buffer was used instead of phosphate buffer because calcium acetate could not completely dissolve in a phosphate ion?rich medium, and an insoluble calcium phosphate was formed. The drug release testing was performed in triplicate.
Analysis of PPN release
The release mechanisms of PPN from the tablets were determined with a power law [19] as shown in Eqns. 1 and 2, as follows: (Mt/M∞)=ktn...(1) and log (Mt/M∞)=n log t + log k...(2) where Mt/M∞ is the fractional PPN release at time t, k is the kinetic constant, and n is the release exponent indicative of the drug?release mechanism. A release exponent of n=0.5 indicates a diffusion?controlled drug release (Fickian diffusion), whereas a release exponent of n=1 corresponds to a polymer?swelling/erosion?controlled release mechanism. Thus, release exponents between these two extreme values indicate so?called anomalous transport, which is a complex transport mechanism that is a mixture of both drug diffusion and the swelling/erosion of the polymer.
The PPN release rate of the tablets was analysed using both zero?order and Higuchi models [20], which can be expressed as Eqns. 3 and 4, respectively, as follows: Q=K0t...(3) and Q=KHt½...(4). Where Q is amount of PPN released, t is time, and K0 and KH are the zero?order and Higuchi release rates, respectively.
Matrix erosion studies
The matrix erosion tests of the tablets containing calcium acetate were performed in a 0.1 M HCl and pH 6.8 Tris buffer with sodium chloride and potassium chloride. The method used in this study was modified from that of a previous report [21,22]. The weighed tablet (Wi) was placed in a basket and subjected to the conditions employed in the release studies described above. Each basket was removed at 1 h, and the morphology of the swollen tablets was viewed using a digital camera (Canon Ixy 920iS, Japan). The baskets were placed in a small beaker and then put in an oven at 50° until the constant weight of the tablet (Wd) was obtained. The percentage of matrix erosion can be calculated using the following equation:
Matrixerosion (%) = 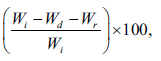 (5)
(5)
where Wr is the mean amount of PPN released at 1 h of the release study. This experiment was performed in triplicate.
Statistical analysis
One?way analysis of variance (ANOVA) with the least significant difference (LSD) test for multiple comparisons and Student’s t?test were used to compare the significantly different results of the thickness, hardness and PPN release parameters of the tablets. All statistical tests were performed using the software SPSS for MS Windows, release 11.5 (SPSS (Thailand) Co. Ltd., Bangkok, Thailand).
Results and Discussion
The matrix tablets containing pure PPN, PPN–MAS physical mixture, or PPN–MAS complexes were successfully prepared using the direct compression method. The LV?HPMC was used as a matrix?forming agent, and the compression pressure applied was 11.0 MPa. The thickness of the tablets obtained was in the range of 5.75?6.44 mm (Table 1). Measuring the different hardness of the tablets found that the PPN tablets showed the lowest hardness. The hardness of the PPN–MAS complex tablets was statistically higher (P<0.05) than that of the PPN–MAS physical mixture tablets. This result suggested that incorporation of MAS into the tablets could increase tablet hardness due to the good compressibility of MAS [9]. Moreover, the particle and surface morphology of MAS (figs. 1a and b) were different than those of the PPN–MAS complex particles (figs. 1c and d). The PPN adsorbed onto the silicate layers of MAS could modify the surface morphology of the PPN–MAS complex particles. The change in surface morphology as well as the particle shape of the complex particles may lead to greater interparticle bonding and interlocking with LV?HPMC particles, resulting in higher tablet hardness.
PPN release profiles of the tablets containing pure PPN, PPN–MAS physical mixture, and PPN–MAS complexes are presented in fig. 2-4. The PPN tablets gave a sustained release profile of PPN in both 0.1 M HCl and pH 6.8 phosphate buffer, whereas an immediately complete dissolution of pure PPN was obtained within 2 min of the test (fig. 2, left panel). The PPN–MAS physical mixture tablets also showed a sustained release of PPN. The PPN–MAS physical mixture gave an incomplete dissolution of PPN, with a 65?75% fast dissolution of PPN, and followed with a decreased amount of PPN dissolved (fig. 3, left panel). This decrease was due to adsorption of PPN with MAS particles [8]. In contrast, the PPN–MAS complexes gave sustained release of PPN after an initial burst release. Incorporation of the PPN–MAS complexes into the matrix tablets could eradicate PPN burst release and could control PPN release (fig. 4, left panel).
The PPN release parameters of all of the tablets are listed in Table 1. The PPN–MAS complex tablets gave the highest release exponent, the n value, which was found to be 0.98 in 0.1 M HCl and 0.89 in pH 6.8 phosphate buffer. These values suggested that the PPN release possibly followed a zero?order release kinetic. The release exponents of the PPN tablets and the PPN–MAS physical mixture tablets were in the range of 0.72?0.75 and 0.62?0.64, respectively, indicating an anomalous transport. The PPN release rates of the tablets were calculated using the zero?order and Higuchi models as shown in Table 1. The PPN release of the PPN tablets and the PPN–MAS complex tablets presented a good fit with a R2 higher than 0.99 when using the zero?order model. However, the Higuchi model gave better fit with the PPN released from the PPN–MAS physical mixture tablet than the zero?order model. The PPN–MAS physical mixture tablets showed the lowest PPN release rate, whereas the highest release rate of PPN was found in the PPN tablets. Additionally, the PPN release rates of the PPN tablets and the PPN–MAS physical mixture tablets in 0.1 M HCl were higher than those in pH 6.8 phosphate buffer. However, the lower PPN release of the PPN–MAS complex tablets in acidic medium was obtained when compared with PPN release in neutral buffer.
The release exponent of the tablets could be used to describe PPN release mechanism from the matrix tablets. The PPN release mechanism model of the PPN tablets is illustrated in fig. 2a, right panel. The PPN powders embedded in the matrix tablets could rapidly dissolve after the tablets were exposed to dissolution medium, owing to the high water solubility of PPN. The PPN molecules diffused through a water?filled channel in the swollen matrix; this process involved tortuosity of the matrix. However, the swollen HPMC matrix could be eroded due to disentanglement and dissolution of HPMC molecules. Thus, the drug release of the PPN tablets was mainly controlled by drug diffusion and a polymer swelling/erosion mechanism. Additionally, the PPN dissolution rate in the medium also involved the drug release mechanism.

Figure 2:Propranolol release profiles and mechanism models of HPMC matrix tablets.
Release profiles are given in left panel and the release mechanisms are provided in right panel. (a) pure PPN. (b) PPN–MAS physical mixture. (c) PPN–MAS complexes. In release profiles: Powders (open symbols); tablets (close symbols) and (?,?) 0.1 M HCl; (?, ?) pH 6.8 phosphate buffer
It was observed that the PPN release rate in 0.1 M HCl was lower than that in pH 6.8 phosphate buffer because the PPN solubility in acidic medium was reported to be 225 mg/ml, whereas the solubility in pH 6.8 phosphate buffer was 130 mg/ml [23], which led to higher PPN release in acidic medium. This finding was in agreement with the previous study [24].
The PPN–MAS physical mixture tablets showed the other process that was involved the PPN release (fig. 2b, right panel). The matrix tablets could absorb water from the surrounding medium, leading to dissolution of PPN powders and swelling of MAS particles in the swollen tablets. Due to the high affinity of a negatively charged MAS with a positively charged PPN [8], an adsorption process of PPN molecules onto the surface of MAS particles occurred. This event resulted in slower PPN release and a lower amount of PPN release at 7 h of the test even though the erosion of the swollen matrix progressed. Therefore, the release exponent of this tablet was smaller than that of the PPN tablets, and the PPN released from this tablet can be described well with the Higuchi model. Moreover, the PPN release rate in acidic medium was also greater than that in pH 6.8 phosphate buffer because of higher PPN solubility and a lower PPN affinity to MAS in acidic medium [8].
In the case of the PPN–MAS complex tablets, the PPN–MAS complex particles embedded in the matrix tablets could absorb cations from the dissolution medium, such as hydrogen and sodium ions. The PPN intercalated in the silicate layers of MAS could be released using a cation exchange process, and this process followed a particle diffusion mechanism within the complex particles [8]. Subsequently, the diffusion of PPN molecules through water?filled channels in the swollen matrix occurred, in which erosion of the swollen matrix also progressed (fig. 2c, right panel). Hence, the particle diffusion?controlled mechanism of the complex particles as drug reservoirs coupled with the drug diffusion and polymer swelling/erosion could control drug release. This outcome resulted in a release exponent close to unity, indicating a zero?order release kinetic of this tablet. However, the PPN release rate in pH 6.8 phosphate buffer was higher than that in acidic medium, in contrast with the PPN tablets and the PPN–MAS physical mixture tablets. To explain this point, a measurement of the matrix erosion of the PPN–MAS complex tablets at 1 h in both media was performed, the results of which were 20.5±2.0% (n=3) for 0.1 M HCl and 35.1±6.7% (n=3) for pH 6.8 phosphate buffer. This result suggested that the faster erosion of the swollen matrix caused a greater PPN release rate in pH 6.8 phosphate buffer. It was also indicated that the drug dissolution process did not involve the PPN release mechanism of the PPN–MAS complex tablets, but this process predominantly controlled the drug released from the PPN tablets and the PPN–MAS physical mixture tablets.
The effect of HPMC viscosity grade on PPN release was investigated in this study. The thickness and hardness of the PPN–MAS complex tablets prepared using different viscosity grades of HPMC and compression pressure at 6.6 MPa are listed in Table 2. The HPMC viscosity grade did not affect the thickness of the tablets prepared. However, the tablet hardness significantly increased (P<0.05) with increasing viscosity grade of HPMC. The use of HV?HPMC presented the highest tablet hardness that was similar to the results of a previous study [25]. This result was due to a lower relative density of HV?HPMC when applying compression pressure [25].
The PPN release profiles of the PPN–MAS complex tablets that were prepared using different grades of HPMC in 0.1 M HCl are shown in fig. 3. The release exponent, the n value, of the tablets seemed to increase when increasing the viscosity grade of HPMC (Table 2). The MV?HPMC and HV?HPMC tablets presented an n value close to unity. However, all of the tablets had a better fit with the zero?order model than with the Higuchi model. The PPN release rate, K0, of the tablets statistically decreased (P<0.05) with increasing HPMC viscosity grade. Generally, increasing the viscosity grade of HPMC produced slower drug release from the HPMC tablets [26]. This result was due to higher viscosity gel barriers created around the tablets when exposed to the dissolution medium. A higher viscosity gel barrier could retard the water absorption rate that in turn affected the ion exchange process of the PPN–MAS complex particles and could reduce drug diffusivity in water?filled channels due to an increase in the tortuosity of swollen matrix. Additionally, slower matrix erosion of the swollen matrix occurred when HV?HPMC was used [27].
| HPMC viscosity grade | Thickness (mm) | Hardness (N) | n | KH(%/min0.5) | K0×10 (%/min) |
|---|---|---|---|---|---|
| LV | 6.22±0.02 | 78.5±5.1 | 0.74±0.03 (R2=0.997) | 4.79±0.17 (R2=0.984) | 3.26±0.01 (R2=0.987) |
| MV | 6.13±0.01 | 288.0±15.0 | 0.92±0.04 (R2=0.993) | 2.62±0.16 (R2=0.936) | 1.20±0.06 (R2=0.998) |
| HV | 6.13±0.01 | 332.4±10.0 | 0.83±0.05 (R2=0.992) | 1.40±0.10 (R2=0.941) | 0.70±0.06 (R2=0.996) |
| HPMC=Hydroxypropylmethylcellulose, LV=Low viscosity, MV=Medium viscosity, HV=High viscosity | |||||
table 2: effect of hpmc viscosity grades on complex-loaded tablets

Figure 3:Effect of viscosity grade of HPMC on propranolol release.
Effect of viscosity grade of HPMC on propranolol release of PPN– MAS complex?loaded HPMC tablets in 0.1 M HCl. (?) LV?HPMC; (?) MV?HPMC; (?) HV?HPMC
The effect of compression pressure on the PPN–MAS complex tablets was also characterised. The thickness of the PPN–MAS complex tablets using LV?HPMC was significantly reduced (P<0.05) when increasing the compression pressure (Table 3). In contrast, an increase in the compression pressure caused a significantly higher hardness of the tablets (P<0.05). These results were similar to those of a previous study [28]. It was indicated that the higher the compression pressure, the lower the porosity of the tablets was obtained, leading to a decrease in tablet thickness. Moreover, HPMC displayed plastic deformation under compression pressure, in which greater compression pressure caused an increase in the interparticle bonding of HPMC particles, resulting in higher tablet hardness [25].
The PPN release of the tablets using different compression pressures in 0.1 M HCl is shown in fig. 4. It can be observed that the compression pressure affected PPN release. The release exponent of the tablets seemed to increase with increasing compression pressure (Table 3). The tablets using 11.0 MPa compression pressure gave a release exponent close to unity, indicative of the zero?order release kinetic, thus leading to a good fit of PPN release with the zero?order model. The PPN release rate, K0, decreased with increasing compression pressure, and a significantly higher PPN release rate of the tablets using 11.0 MPa compression pressure was found (P<0.05) when compared with those using 6.6 MPa compression pressure. This finding was in contrast with that of the previous study in which the compression pressure had little influence on the drug release from HPMC tablets [28?31]. The matrix erosion of the tablets was also investigated, as shown in Table 3. It was shown that tablets using different compression pressures were similar with regards to matrix erosion. This result suggested that higher tablet hardness did not influence the water absorption and swelling processes of the tablets when exposed to dissolution medium because of a change in the PPN–MAS complex particles embedded in the matrix tablets under compression pressure. Previously, the drug–MAS complex particles without other excipients could be compressed as a tablet that provided a very high hardness [9]. This result led to slower drug release of the drug–MAS complex tablets when compared with the drug?MAS complex particles. Thus, the PPN–MAS complex particles could possibly deform under higher compression pressure, which may cause a slower PPN release within the deformed complex particles. Hence, this finding suggested that the use of the PPN–MAS complexes as drug reservoirs in the HPMC tablets was sensitive to compression pressure as opposed to the HPMC tablets containing pure PPN.

Figure 4:Effect of compression pressure on propranolol release.
Effect of compression pressure on propranolol (PPN) release of PPN–MAS complex?loaded HPMC tablets in 0.1 M HCl (Δ) 6.6; (?) 8.8; (?) 11.0 MPa compression pressure
The PPN–MAS complex tablets that incorporated various amounts of calcium acetate were prepared using LV?HPMC and 6.6 MPa compression pressure. The thickness of the PPN–MAS complex tablets tended to decrease when 150 mg of calcium acetate was incorporated (Table 4). In contrast, the tablet hardness was significantly reduced (P<0.05) when adding 50 and 100 mg of calcium acetate, whereas a statistical increase of tablet hardness was found (P<0.05) in the tablets with 150 mg of calcium acetate, compared with the control tablets. These results suggested that calcium acetate could reduce interparticle bonding of LV?HPMC when adding lower levels of calcium acetate. However, the highest amount (150 mg) of calcium acetate caused an increase in tablet hardness because calcium acetate may have a good compressibility and may form interparticle bonding amongst itself; this bonding could be observed from a reduction of tablet thickness.
The PPN release profiles of the PPN–MAS complex tablets containing various amounts of calcium acetate in 0.1 M HCl and pH 6.8 Tris buffer are shown in fig. 5a and b, respectively. Using 0.1 M HCl, the release exponent, the n value of the tablets was not affected by the incorporation of calcium acetate; these values were over the range 0.67?0.75 (Table 4). The matrix erosion of the swollen tablets statistically increased (P<0.05) when calcium acetate was added, but did not relate to the increase of calcium acetate amount. This result can be confirmed by using a photo image of the swollen tablets at 1 h of the release testing as presented in fig. 6. The similarity in morphology of the swollen tablets with or without calcium acetate was observed. However, the PPN release rate, K0, of the tablets with calcium acetate was significantly higher (P<0.05) than that of the control tablets. The greater the calcium acetate incorporated, the higher the PPN release rate was found. In addition to the results in acidic medium, the n value of the PPN release in pH 6.8 Tris buffer was close to unity when calcium acetate was incorporated into the tablets (Table 4). The swollen tablets showed a higher percentage of matrix erosion when incorporating calcium acetate (Table 4). The morphology of the swollen tablets with calcium acetate was changed (fig. 6), indicating that the incorporation of calcium acetate promoted matrix erosion of the swollen tablets in pH 6.8 Tris buffer. It was also observed that the matrix erosion of the tablets in pH 6.8 Tris buffer was greater than that in 0.1 M HCl. This result led to higher PPN release rate in pH 6.8 Tris buffer when compared with using acidic medium.

Figure 5:Effect of calcium acetate amount on propranolol release.
Effect of calcium acetate amount on propranolol (PPN) release of PPN–MAS complex?loaded HPMC tablets in 0.1 M HCl (a) and pH 6.8 Tris buffer (b). (?) 0; (?); 50; (?) 100; (?) 150 mg calcium acetate.

Figure 6:Morphology of swollen matrix of PPN–MAS complex tablets.
Photo images of swollen matrix morphology of PPN–MAS complex tablets containing various amounts of calcium acetate in 0.1 M HCl (row 1) and pH 6.8 Tris buffer (row 2) at 1 h of release testing
Calcium acetate could be dissolved, thus providing calcium ions in the swollen matrix tablets. The divalent calcium ions could accelerate an ion exchange process of the PPN–MAS complexes, resulting in higher release of PPN from the site of adsorption on the silicate layers of MAS. This occurrence led to higher release rate of PPN from the tablets. However, the effect of calcium ions could be clearly observed when the matrix erosion of the swollen HPMC tablets occurred slowly and calcium ions had sufficient time for diffusion into the complex particles for ion exchange process. These phenomena could be found when using 0.1 M HCl as a dissolution medium. An increase in the calcium acetate amount did not promote matrix erosion of the swollen tablets, which would subsequently have allowed the liberation of calcium ions that in turn could accelerate the ion exchange process and result in a higher PPN release rate. In contrast to the use of pH 6.8 Tris buffer, the matrix erosion of the swollen tablets progressed rapidly such that the action of calcium ions could not completely occur. Thus, the higher release rate of the PPN–MAS complex tablets in pH 6.8 Tris buffer was primarily controlled by the matrix erosion mechanism.
| Compression pressure (MPa) | Thickness (mm) | Hardness (N) | n | K H (%/min0.5) | K0 ×10 (%/min) ) | ME (% |
|---|---|---|---|---|---|---|
| 6.6 | 6.24±0.02 | 78.5±5.1 | 0.74±0.03 (R2=0.997) 4.79±0.17 (R2=0.984) | 3.26±0.01 (R2=0.987) | 20.7±1.9 | |
| 8.8 | 6.06±0.02 | 124.5±6.1 | 0.75±0.10 (R2=0.980) 4.31±0.42 (R2=0.960) | 3.00±0.21 (R2=0.990) | 18.7±0.2 | |
| 11.0 | 5.75±0.03 | 179.1±4.4 | 0.98±0.02 (R2=0.993) | 3.91±0.60 (R2=0.959) | 2.70±0.44 (R2=0.999) | 20.5±2.0 |
| ME=Matrix erosion | ||||||
Table 3: Effect of compressure pressures on complex-loaded tablets
| CA (mg) | Thickness | Hardness | 0.1 M HCl | pH 6.8 Tris buffer | ||||
|---|---|---|---|---|---|---|---|---|
| (mm) | (N) | n | K0×10 (%/min) | ME (%) | n | K0×10 (%/min) | ME (%) | |
| 0 | 6.22±0.02 | 78.5±5.1 | 0.74±0.03 (R2=0.997) 3.26±0.01 (R2=0.987) 20.7±1.9 | 0.79±0.03 (R2=0.990) 3.38±0.02 (R2=0.972) 39.0±0.5 | ||||
| 50 | 6.13±0.01 | 51.3±4.4 | 0.75±0.03 (R2=0.996) 4.07±0.05 (R2=0.991) 35.0±1.4 | 0.79±0.05 (R2=0.997) 5.29±0.03 (R2=0.990) 51.2±3.3 | ||||
| 100 | 6.23±0.01 | 69.0±2.5 | 0.67±0.04 (R2=0.999) 4.10±0.01 (R2=0.992) 31.6±3.6 | 0.93±0.05 (R2=0.997) 3.49±0.03 (R2=0.998) 41.8±5.1 | ||||
| 150 | 6.06±0.02 | 93.8±5.9 | 0.74±0.06 (R2=0.993) | 4.45±0.02 (R2=0.995) | 33.3±6.1 | 0.82±0.03 (R2=0.991) | 4.73±0.02 (R2=0.992) | 53.9±2.5 |
Table 4: Effect of calcium acetate amounts on complex-loaded tablets.
In conclusion, the PPN–MAS complex tablets were successfully prepared using the direct compression method, and HPMC was used as a matrix?forming agent. The PPN–MAS complex tablets gave higher tablet hardness than the tablets containing pure PPN or the PPN–MAS physical mixture. The PPN release from the PPN–MAS complex tablets followed a zero?order release kinetic because the release of PPN was controlled by many mechanisms, such as a cation exchange process, a particle diffusion?controlled mechanism of the complex particles, drug diffusion through water?filled channel in the matrix and polymer swelling/erosion of HPMC. The PPN release rate of the PPN–MAS complex tablet decreased with increasing HPMC viscosity grade. The higher compression pressure used could slow the PPN release from these tablets. Additionally, calcium salt incorporated into the tablets could accelerate PPN release, particularly in acidic medium, because calcium ion could exchange with PPN intercalated in the silicate layers of MAS. These findings suggest that PPN–MAS intercalated complexes can be used as drug reservoirs in polymeric matrix tablets intended for modifying drug release in oral drug delivery systems.
Acknowledgment
The authors would like to thank the Thailand Research Fund (Bangkok, Thailand) for research funding (Grant No. RSA5280013). We are very pleased to acknowledge the Faculty of Pharmaceutical Sciences, Khon Kaen University (Khon Kaen, Thailand), for technical support.
References
- Sinko PJ. Complexation and protein binding, Martin’s Physical Pharmacy and Pharmaceutical Sciences. 6th ed. Philadelphia: Lippincott Williams and Wilkins; 2011. p. 197-222.
- Velde B. Tools, Introduction to Clay Minerals. London: Chapman and Hall; 1992. p. 12-36.
- Murray HH. Traditional and new applications for kaolin, smectite, and palygorskite: A general overview. Appl Clay Sci 2000;17:207-21.
- Kibbe HA. Magnesium aluminium silicate, Handbook of Pharmaceutical Excipients. 3rd ed. Washington: American Pharmaceutical Association; 2000. p. 295-8.
- Alexandre M, Dubois P. Polymer-layered silicate nanocomposites: Preparation, properties and uses of a new class of materials. Mater SciEng 2000;28:1-63.
- Nunes CD, Vaz PD, Fernandes AC, Ferreira P, Romão CC, Calhorda MJ. Loading and delivery of sertraline using inorganic micro and mesoporous materials. Eur J Pharm Biopharm 2007;66:357-65.
- Pongjanyakul T, Khunawattanakul W, Puttipipatkhachorn S. Physicochemical characterizations and release studies of nicotine-magnesium aluminum silicate complexes. Appl Clay Sci 2009;44:242-50.
- Rojtanatanya S, Pongjanyakul T. Propranolol-magnesium aluminum silicate complex dispersions and particles: Characterization and factors influencing drug release. Int J Pharm 2010;383:106-15.
- Kanjanabat S, Pongjanyakul T. Preparation and characterization of nicotine-magnesium aluminum silicate complex-loaded sodium alginate matrix tablets for buccal delivery. AAPS PharmSciTech 2011;12:683-92.
- Dollery SC. Propranolol (hydrochloride), Therapeutic Drugs. Edinburgh: Churchill Livingstone; 1991. p. 272-8.
- Gil EC, Colarte AI, Bataille B, Pedraz JL, Rodríguez F, Heinämäki J. Development and optimization of a novel sustained-release dextran tablet formulation for propranolol hydrochloride. Int J Pharm 2006;317:32-9.
- Sahoo J, Murthy PN, Biswal S, Sahoo SK, Mahapatra AK. Comparative study of propranolol hydrochloride release from matrix tablets with Kollidon SR or hydroxypropyl methylcellulose. AAPS PharmSciTech 2008;9:577-82.
- Paker-Leggs S, Neau SH. Pellet characteristics and drug release when the form of propranolol is fixed as moles or mass in formulations for extruded and spheronizedCarbopol-containing pellets. Int J Pharm 2009;369:96-104.
- Chakraborty S, Khandai M, Sharma A, PatraChN, Patro VJ, Sen KK. Effects of drug solubility on the release kinetics of water soluble and insoluble drugs from HPMC based matrix formulations. Acta Pharm 2009;59:313-23.
- Basak SC, Srinivasa Rao Y, Manavalan R, Rama Rao P. Controlled release HPMC matrix tablets of propranolol hydrochloride. Ind J Pharm Sci 2004;66:827-30.
- Basak SC, Jayakumar Reddy BM, Lucas Mani KP. Formulation and release behaviour of sustained release ambroxol hydrochloride HPMC matrix tablet. Indian J Pharm Sci 2006;68:594-8.
- Mughal MA, Iqbal Z, Neau SH. Guar gum, xanthan gum, and HPMC can define release mechanisms and sustain release of propranolol hydrochloride. AAPS PharmSciTech 2011;12:77-87.
- Notari RE. Biopharmaceutics, Biopharmaceutics and Clinical Pharmacokinetics. New York: Marcel Dekker; 1987. p. 130-220.
- Siepmann J, Siepmann F. Mathematical modeling of drug release from lipid dosage forms. Int J Pharm 2011;418:42-53.
- Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci 2001;13:123-33.
- Sutananta W, Craig DQ, Newton JM. An evaluation of the mechanisms of drug release from glyceride bases. J Pharm Pharmacol 1995;47:182-7.
- Pongjanyakul T, Medlicott NJ, Tucker IG. Melted glycerylpalmitostearate (GPS) pellets for protein delivery. Int J Pharm 2004;271:53-62.
- Takka S, Rajbhandari S, Sakr A. Effect of anionic polymers on the release of propranolol hydrochloride from matrix tablets. Eur J Pharm Biopharm 2001;52:75-82.
- Colombo P, Bettini R, Massimo G, Catellani PL, Santi P, Peppas NA. Drug diffusion front movement is important in drug release control from swellable matrix tablets. J Pharm Sci 1995;84:991-7.
- Nokhodchi A, Ford JL, Rowe PH, Rubinstein MH. The effects of compression rate and force on the compaction properties of different viscosity grades of hydroxypropyl methylcellulose 2208. Int J Pharm 1996;129:21-31.
- Campos-Aldrete ME, Villafuerte-Robles L. Influence of the viscosity grade and the particle size of HPMC on metronidazole release from matrix tablets. Eur J Pharm Biopharm 1997;43:173-8.
- Siepmann J, Peppas NA. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv Drug Deliv Rev 2001;48:139-57.
- Velasco MV, Ford JL, Rowe P, Rajabi-Siahboomi AR. Influence of drug: Hydroxypropylmethylcellulose ratio, drug and polymer particle size and compression force on the release of diclofenac sodium from HPMC tablets. J Control Release 1999;57:75-85.
- Ford JL, Rubinstein MH, Hogan JE. Propranolol hydrochloride and aminophylline release from matrix tablets containing hydroxypropyl methylcellulose. Int J Pharm 1985;24:339-50.
- Dahl TC, Calderwood T, Bormeth A, Trimble K, Piepmeir E. Influence of physio-chemical properties of hydroxypropyl methylcellulose in naproxen release from sustained release matrix tablets. J Control Release 1990;14:1-10.
- Liu CH, Kao YH, Chen SC, Sokoloski TD, Sheu MT. In vitro and in vivo studies of the diclofenac sodium controlled-release matrixtablets. JPharm Pharmacol 1995;47:360-4.





